InVivoMAb anti-mouse CD19
Product Details
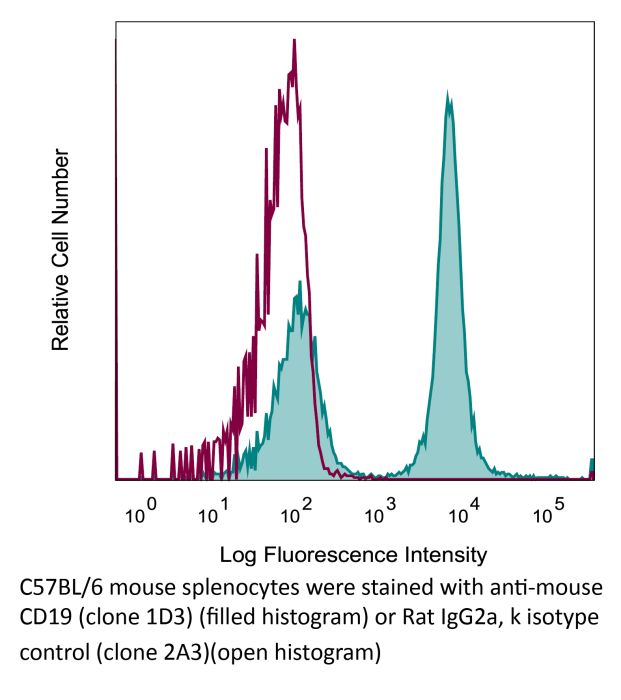
The 1D3 monoclonal antibody reacts with mouse CD19, a B cell-specific 95 kDa transmembrane glycoprotein of the immunoglobulin superfamily. CD19 contains two extracellular immunoglobulin-like domains and an extensive cytoplasmic tail. It functions as a positive regulator of B-cell receptor signaling in conjunction with CD21 and CD81. CD19 is highly expressed in most lymphomas and leukemias including some early B-cell malignancies that do not express CD20. For these reasons CD19 is quickly becoming an attractive alternative target for the immunotherapy of lymphoproliferative disorders. The 1D3 antibody has been reported to deplete B cells however, B cell depletion often requires treatment with a combination of 1D3, anti-mouse B220 (clone RA3.3A1/6.1), anti-mouse CD22 (clone Cy34.1), and anti-rat κ light chain (clone MAR 18.5) antibodies. Please see the provided references for protocol details.Specifications
| Isotype | Rat IgG2a, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb rat IgG2a isotype control, anti-trinitrophenol |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | K562 cells expressing the extracellular domain of mouse CD19 |
| Reported Applications |
in vivo B cell depletion in vivo CD19 neutralization in vitro B cell negative selection Flow cytometry |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
<2EU/mg (<0.002EU/μg) Determined by LAL gel clotting assay |
| Purity |
>95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_10949187 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
Recommended Products
-

Recommended Isotype Control(s)
InVivoMAb rat IgG2a isotype control, anti-trinitrophenol
-

Recommended Dilution Buffer
InVivoPure pH 7.0 Dilution Buffer
in vivo B cell depletion
Sawen, P., et al. (2016). "Mitotic History Reveals Distinct Stem Cell Populations and Their Contributions to Hematopoiesis" Cell Rep 14(12): 2809-2818. PubMed
Homeostasis of short-lived blood cells is dependent on rapid proliferation of immature precursors. Using a conditional histone 2B-mCherry-labeling mouse model, we characterize hematopoietic stem cell (HSC) and progenitor proliferation dynamics in steady state and following several types of induced stress. HSC proliferation following HSC transplantation into lethally irradiated mice is fundamentally different not only from native hematopoiesis but also from other stress contexts. Whereas transplantation promoted sustained, long-term proliferation of HSCs, both cytokine-induced mobilization and acute depletion of selected blood cell lineages elicited very limited recruitment of HSCs to the proliferative pool. By coupling mCherry-based analysis of proliferation history with multiplex gene expression analyses on single cells, we have found that HSCs can be stratified into four distinct subtypes. These subtypes have distinct molecular signatures and differ significantly in their reconstitution potentials, showcasing the power of tracking proliferation history when resolving functional heterogeneity of HSCs.
Flow Cytometry
Becker, A. M., et al. (2015). "ADAM17 limits the expression of CSF1R on murine hematopoietic progenitors" Exp Hematol 43(1): 44-52 e41-43. PubMed
All-lymphoid progenitors (ALPs) yield few myeloid cells in vivo, but readily generate such cells in vitro. The basis for this difference remains unknown. We hypothesized that ALPs limit responsiveness to in vivo concentrations of myeloid-promoting cytokines by reducing expression of the corresponding receptors, potentially through posttranscriptional mechanisms. Consistent with such a mechanism, ALPs express higher levels of CSF1R transcripts than their upstream precursors, yet show limited cell-surface protein expression of colony-stimulating factor 1 receptor (CSF1R). All-lymphoid progenitors and other hematopoietic progenitors deficient in A disintegrin and metalloproteinase domain 17 (ADAM17), display elevated cell surface CSF1R expression. ADAM17(-/-) ALPs, however, fail to yield myeloid cells upon transplantation into irradiated recipients. Moreover, ADAM17(-/-) ALPs yield fewer macrophages in vitro than control ALPs at high concentrations of macrophage colony stimulating factor. Mice with hematopoietic-specific deletion of ADAM17 have normal numbers of myeloid and lymphoid progenitors and mature cells in vivo. These data demonstrate that ADAM17 limits CSF1R protein expression on hematopoietic progenitors, but that compensatory mechanisms prevent elevated CSF1R levels from altering lymphoid progenitor potential.
in vivo B cell depletion
Carmi, Y., et al. (2015). "Allogeneic IgG combined with dendritic cell stimuli induce antitumour T-cell immunity" Nature 521(7550): 99-104. PubMed
Whereas cancers grow within host tissues and evade host immunity through immune-editing and immunosuppression, tumours are rarely transmissible between individuals. Much like transplanted allogeneic organs, allogeneic tumours are reliably rejected by host T cells, even when the tumour and host share the same major histocompatibility complex alleles, the most potent determinants of transplant rejection. How such tumour-eradicating immunity is initiated remains unknown, although elucidating this process could provide the basis for inducing similar responses against naturally arising tumours. Here we find that allogeneic tumour rejection is initiated in mice by naturally occurring tumour-binding IgG antibodies, which enable dendritic cells (DCs) to internalize tumour antigens and subsequently activate tumour-reactive T cells. We exploited this mechanism to treat autologous and autochthonous tumours successfully. Either systemic administration of DCs loaded with allogeneic-IgG-coated tumour cells or intratumoral injection of allogeneic IgG in combination with DC stimuli induced potent T-cell-mediated antitumour immune responses, resulting in tumour eradication in mouse models of melanoma, pancreas, lung and breast cancer. Moreover, this strategy led to eradication of distant tumours and metastases, as well as the injected primary tumours. To assess the clinical relevance of these findings, we studied antibodies and cells from patients with lung cancer. T cells from these patients responded vigorously to autologous tumour antigens after culture with allogeneic-IgG-loaded DCs, recapitulating our findings in mice. These results reveal that tumour-binding allogeneic IgG can induce powerful antitumour immunity that can be exploited for cancer immunotherapy.
in vitro B cell negative selection
Liu, B., et al. (2015). "Collaborative interactions between type 2 innate lymphoid cells and antigen-specific CD4+ Th2 cells exacerbate murine allergic airway diseases with prominent eosinophilia" J Immunol 194(8): 3583-3593. PubMed
Type-2 innate lymphoid cells (ILC2s) and the acquired CD4(+) Th2 and Th17 cells contribute to the pathogenesis of experimental asthma; however, their roles in Ag-driven exacerbation of chronic murine allergic airway diseases remain elusive. In this study, we report that repeated intranasal rechallenges with only OVA Ag were sufficient to trigger airway hyperresponsiveness, prominent eosinophilic inflammation, and significantly increased serum OVA-specific IgG1 and IgE in rested mice that previously developed murine allergic airway diseases. The recall response to repeated OVA inoculation preferentially triggered a further increase of lung OVA-specific CD4(+) Th2 cells, whereas CD4(+) Th17 and ILC2 cell numbers remained constant. Furthermore, the acquired CD4(+) Th17 cells in Stat6(-/-)/IL-17-GFP mice, or innate ILC2s in CD4(+) T cell-ablated mice, failed to mount an allergic recall response to OVA Ag. After repeated OVA rechallenge or CD4(+) T cell ablation, the increase or loss of CD4(+) Th2 cells resulted in an enhanced or reduced IL-13 production by lung ILC2s in response to IL-25 and IL-33 stimulation, respectively. In return, ILC2s enhanced Ag-mediated proliferation of cocultured CD4(+) Th2 cells and their cytokine production, and promoted eosinophilic airway inflammation and goblet cell hyperplasia driven by adoptively transferred Ag-specific CD4(+) Th2 cells. Thus, these results suggest that an allergic recall response to recurring Ag exposures preferentially triggers an increase of Ag-specific CD4(+) Th2 cells, which facilitates the collaborative interactions between acquired CD4(+) Th2 cells and innate ILC2s to drive the exacerbation of a murine allergic airway diseases with an eosinophilic phenotype.
in vivo CD19 neutralization, Flow Cytometry
Dai, M., et al. (2015). "Curing mice with large tumors by locally delivering combinations of immunomodulatory antibodies" Clin Cancer Res 21(5): 1127-1138. PubMed
PURPOSE: Immunomodulatory mAbs can treat cancer, but cures are rare except for small tumors. Our objective was to explore whether the therapeutic window increases by combining mAbs with different modes of action and injecting them into tumors. EXPERIMENTAL DESIGN: Combinations of mAbs to CD137/PD-1/CTLA-4 or CD137/PD-1/CTLA-4/CD19 were administrated intratumorally to mice with syngeneic tumors (B16 and SW1 melanoma, TC1 lung carcinoma), including tumors with a mean surface of approximately 80 mm(2). Survival and tumor growth were assessed. Immunologic responses were evaluated using flow cytometry and qRT-PCR. RESULTS: More than 50% of tumor-bearing mice had complete regression and long-term survival after tumor injection with mAbs recognizing CD137/PD-1/CTLA-4/CD19 with similar responses in three models. Intratumoral injection was more efficacious than intraperitoneal injection in causing rejection also of untreated tumors in the same mice. The three-mAb combination could also induce regression, but was less efficacious. There were few side effects, and therapy-resistant tumors were not observed. Transplanted tumor cells rapidly caused a Th2 response with increased CD19 cells. Successful therapy shifted this response to the Th1 phenotype with decreased CD19 cells and increased numbers of long-term memory CD8 effector cells and T cells making IFNgamma and TNFalpha. CONCLUSIONS: Intratumoral injection of mAbs recognizing CD137/PD-1/CTLA-4/CD19 can eradicate established tumors and reverse a Th2 response with tumor-associated CD19 cells to Th1 immunity, whereas a combination lacking anti-CD19 is less effective. There are several human cancers for which a similar approach may provide clinical benefit.
in vitro B cell negative selection
Bouffi, C., et al. (2015). "Transcription Factor Repertoire of Homeostatic Eosinophilopoiesis" J Immunol 195(6): 2683-2695. PubMed
The production of mature eosinophils (Eos) is a tightly orchestrated process with the aim to sustain normal Eos levels in tissues while also maintaining low numbers of these complex and sensitive cells in the blood. To identify regulators of homeostatic eosinophilopoiesis in mice, we took a global approach to identify genome-wide transcriptome and epigenome changes that occur during homeostasis at critical developmental stages, including Eos-lineage commitment and lineage maturation. Our analyses revealed a markedly greater number of transcriptome alterations associated with Eos maturation (1199 genes) than with Eos-lineage commitment (490 genes), highlighting the greater transcriptional investment necessary for differentiation. Eos-lineage-committed progenitors (EoPs) were noted to express high levels of granule proteins and contain granules with an ultrastructure distinct from that of mature resting Eos. Our analyses also delineated a 976-gene Eos-lineage transcriptome that included a repertoire of 56 transcription factors, many of which have never previously been associated with Eos. EoPs and Eos, but not granulocyte-monocyte progenitors or neutrophils, expressed Helios and Aiolos, members of the Ikaros family of transcription factors, which regulate gene expression via modulation of chromatin structure and DNA accessibility. Epigenetic studies revealed a distinct distribution of active chromatin marks between genes induced with lineage commitment and genes induced with cell maturation during Eos development. In addition, Aiolos and Helios binding sites were significantly enriched in genes expressed by EoPs and Eos with active chromatin, highlighting a potential novel role for Helios and Aiolos in regulating gene expression during Eos development.
in vivo B cell depletion
Guo, Z., et al. (2013). "Combined TIM-3 blockade and CD137 activation affords the long-term protection in a murine model of ovarian cancer" J Transl Med 11: 215. PubMed
BACKGROUND: T-cell immunoglobulin and mucin domain 3 (TIM-3) is known as a negative immune regulator and emerging data have implicated TIM-3 a pivotal role in suppressing antitumor immunity. The co-stimulatory receptor CD137 is transiently upregulated on T-cells following activation and increases their proliferation and survival when engaged. Although antagonistic anti-TIM-3 or agonistic anti-CD137 antibodies can promote the rejection of several murine tumors, some poorly immunogenic tumors were refractory to this treatment. In this study, we sought to evaluate whether combined TIM-3 blockade and CD137 activation would significantly improve the immunotherapy in the murine ID8 ovarian cancer model. METHODS: Mice with established ID8 tumor were intraperitoneally injected with single or combined anti-TIM-3/CD137 monoclonal antibody (mAb); mice survival was recorded, the composition and gene expression of tumor-infiltrating immune cells in these mice was analyzed by flow cytometry and quantitative RT-PCR respectively, and the function of CD8(+) cells was evaluated by ELISA and cytotoxicity assay. RESULTS: Either anti-TIM-3 or CD137 mAb alone, although effective in 3 days established tumor, was unable to prevent tumor progression in mice bearing 10 days established tumor, however, combined anti-TIM-3/CD137 mAb significantly inhibited the growth of these tumors with 60% of mice tumor free 90 days after tumor inoculation. Therapeutic efficacy was associated with a systemic immune response with memory and antigen specificity, required CD4(+) cells and CD8(+) cells. The 2 mAb combination increased CD4(+) and CD8(+) cells and decreased immunosuppressive CD4(+)FoxP3(+) regulatory T (Treg) cells and CD11b(+)Gr-1(+) myeloid suppressor cells (MDSC) at tumor sites, giving rise to significantly elevated ratios of CD4(+) and CD8(+) cells to Treg and MDSC; This is consistent with biasing local immune response towards an immunostimulatory Th1 type and is further supported by quantitative RT-PCR data showing the increased Th1-associated genes by anti-TIM-3/CD137 treatment. The increased CD8(+) T cells produced high level of IFN-gamma upon tumor antigen stimulation and displayed antigen-specific cytotoxic activity. CONCLUSIONS: To our knowledge, this is the first report investigating the effects of anti-TIM-3/CD137 combined mAb in a murine ovarian cancer model, and our results may aid the design of future trials for ovarian cancer immunotherapy.
in vivo CD19 neutralization, Flow Cytometry
Dai, M., et al. (2013). "Long-lasting complete regression of established mouse tumors by counteracting Th2 inflammation" J Immunother 36(4): 248-257. PubMed
40% of mice with SW1 tumors remained healthy >150 days after last treatment and are probably cured. Therapeutic efficacy was associated with a systemic immune response with memory and antigen specificity, required CD4 cells and involved CD8 cells and NK cells to a less extent. The 3 mAb combination significantly decreased CD19 cells at tumor sites, increased IFN-gamma and TNF-alpha producing CD4 and CD8 T cells and mature CD86 dendritic cells (DC), and it increased the ratios of effector CD4 and CD8 T cells to CD4Foxp3 regulatory T (Treg) cells and to CD11bGr-1 myeloid suppressor cells (MDSC). This is consistent with shifting the tumor microenvironment from an immunosuppressive Th2 to an immunostimulatory Th1 type and is further supported by PCR data. Adding an anti-CD19 mAb to the 3 mAb combination in the SW1 model further increased therapeutic efficacy. Data from ongoing experiments show that intratumoral injection of a combination of mAbs to CD137PD-1CTLA4CD19 can induce complete regression and dramatically prolong survival also in the TC1 carcinoma and B16 melanoma models, suggesting that the approach has general validity.”}” data-sheets-userformat=”{“2″:14851,”3”:{“1″:0},”4”:{“1″:2,”2″:16777215},”12″:0,”14”:{“1″:2,”2″:1521491},”15″:”Roboto, sans-serif”,”16″:12}”>Mice with intraperitoneal ID8 ovarian carcinoma or subcutaneous SW1 melanoma were injected with monoclonal antibodies (mAbs) to CD137PD-1CTLA4 7-15 days after tumor initiation. Survival of mice with ID8 tumors tripled and >40% of mice with SW1 tumors remained healthy >150 days after last treatment and are probably cured. Therapeutic efficacy was associated with a systemic immune response with memory and antigen specificity, required CD4 cells and involved CD8 cells and NK cells to a less extent. The 3 mAb combination significantly decreased CD19 cells at tumor sites, increased IFN-gamma and TNF-alpha producing CD4 and CD8 T cells and mature CD86 dendritic cells (DC), and it increased the ratios of effector CD4 and CD8 T cells to CD4Foxp3 regulatory T (Treg) cells and to CD11bGr-1 myeloid suppressor cells (MDSC). This is consistent with shifting the tumor microenvironment from an immunosuppressive Th2 to an immunostimulatory Th1 type and is further supported by PCR data. Adding an anti-CD19 mAb to the 3 mAb combination in the SW1 model further increased therapeutic efficacy. Data from ongoing experiments show that intratumoral injection of a combination of mAbs to CD137PD-1CTLA4CD19 can induce complete regression and dramatically prolong survival also in the TC1 carcinoma and B16 melanoma models, suggesting that the approach has general validity.
in vitro B cell negative selection, Flow Cytometry
De Obaldia, M. E., et al. (2013). "T cell development requires constraint of the myeloid regulator C/EBP-alpha by the Notch target and transcriptional repressor Hes1" Nat Immunol 14(12): 1277-1284. PubMed
Notch signaling induces gene expression of the T cell lineage and discourages alternative fate outcomes. Hematopoietic deficiency in the Notch target Hes1 results in severe T cell lineage defects; however, the underlying mechanism is unknown. We found here that Hes1 constrained myeloid gene-expression programs in T cell progenitor cells, as deletion of the myeloid regulator C/EBP-alpha restored the development of T cells from Hes1-deficient progenitor cells. Repression of Cebpa by Hes1 required its DNA-binding and Groucho-recruitment domains. Hes1-deficient multipotent progenitor cells showed a developmental bias toward myeloid cells and dendritic cells after Notch signaling, whereas Hes1-deficient lymphoid progenitor cells required additional cytokine signaling for diversion into the myeloid lineage. Our findings establish the importance of constraining developmental programs of the myeloid lineage early in T cell development.
Flow Cytometry
Purtha, W. E., et al. (2012). "Spontaneous mutation of the Dock2 gene in Irf5-/- mice complicates interpretation of type I interferon production and antibody responses" Proc Natl Acad Sci U S A 109(15): E898-904. PubMed
Genome-wide studies have identified associations between polymorphisms in the IFN regulatory factor-5 (Irf5) gene and a variety of human autoimmune diseases. Its functional role in disease pathogenesis, however, remains unclear, as studies in Irf5(-/-) mice have reached disparate conclusions regarding the importance of this transcription factor in type I IFN production and antibody responses. We identified a spontaneous genomic duplication and frameshift mutation in the guanine exchange factor dedicator of cytokinesis 2 (Dock2) that has arisen in at least a subset of circulating Irf5(-/-) mice and inadvertently been bred to homozygosity. Retroviral expression of DOCK2, but not IRF-5, rescued defects in plasmacytoid dendritic cell and B-cell development, and Irf5(-/-) mice lacking the mutation in Dock2 exhibited normal plasmacytoid dendritic cell and B-cell development, largely intact type I IFN responses, and relatively normal antibody responses to viral infection. Thus, confirmation of the normal Dock2 genotype in circulating Irf5(-/-) mice is warranted, and our data may partly explain conflicting results in this field.
in vivo B cell depletion
Keren, Z., et al. (2011). "B-cell depletion reactivates B lymphopoiesis in the BM and rejuvenates the B lineage in aging" Blood 117(11): 3104-3112. PubMed
Aging is associated with a decline in B-lymphopoiesis in the bone marrow and accumulation of long-lived B cells in the periphery. These changes decrease the body’s ability to mount protective antibody responses. We show here that age-related changes in the B lineage are mediated by the accumulating long-lived B cells. Thus, depletion of B cells in old mice was followed by expansion of multipotent primitive progenitors and common lymphoid progenitors, a revival of B-lymphopoiesis in the bone marrow, and generation of a rejuvenated peripheral compartment that enhanced the animal’s immune responsiveness to antigenic stimulation. Collectively, our results suggest that immunosenescence in the B-lineage is not irreversible and that depletion of the long-lived B cells in old mice rejuvenates the B-lineage and enhances immune competence.
Flow Cytometry
Purtha, W. E., et al. (2011). "Memory B cells, but not long-lived plasma cells, possess antigen specificities for viral escape mutants" J Exp Med 208(13): 2599-2606. PubMed
Memory B cells (MBCs) and long-lived plasma cells (LLPCs) persist after clearance of infection, yet the specific and nonredundant role MBCs play in subsequent protection is unclear. After resolution of West Nile virus infection in mice, we demonstrate that LLPCs were specific for a single dominant neutralizing epitope, such that immune serum poorly inhibited a variant virus that encoded a mutation at this critical epitope. In contrast, a large fraction of MBC produced antibody that recognized both wild-type (WT) and mutant viral epitopes. Accordingly, antibody produced by the polyclonal pool of MBC neutralized WT and variant viruses equivalently. Remarkably, we also identified MBC clones that recognized the mutant epitope better than the WT protein, despite never having been exposed to the variant virus. The ability of MBCs to respond to variant viruses in vivo was confirmed by experiments in which MBCs were adoptively transferred or depleted before secondary challenge. Our data demonstrate that class-switched MBC can respond to variants of the original pathogen that escape neutralization of antibody produced by LLPC without a requirement for accumulating additional somatic mutations.