Quote
You have no items in your quote cart.
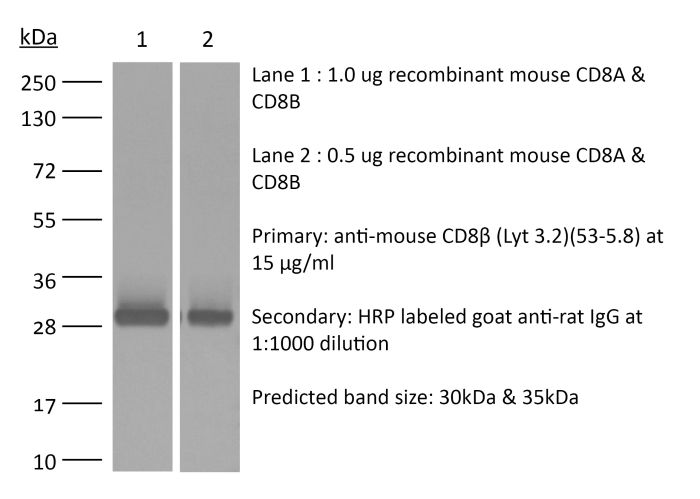
| Isotype | Rat IgG1, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb rat IgG1 isotype control, anti-horseradish peroxidase |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Mouse thymus or spleen |
| Reported Applications |
in vivo CD8+ T cell depletion in vitro CD8 blockade Immunofluorescence |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
<2EU/mg (<0.002EU/μg) Determined by LAL gel clotting assay |
| Purity |
>95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_2687706 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |

Recommended Isotype Control(s)

Recommended Dilution Buffer
In the murine model of cerebral malaria caused by P. berghei ANKA (PbA), parasite-specific CD8+ T cells directly induce pathology and have long been hypothesized to kill brain endothelial cells that have internalized PbA antigen. We previously reported that brain microvessel fragments from infected mice cross-present PbA epitopes, using reporter cells transduced with epitope-specific T cell receptors. Here, we confirm that endothelial cells are the population responsible for cross-presentation in vivo, not pericytes or microglia. PbA antigen cross-presentation by primary brain endothelial cells in vitro confers susceptibility to killing by CD8+ T cells from infected mice. IFNgamma stimulation is required for brain endothelial cross-presentation in vivo and in vitro, which occurs by a proteasome- and TAP-dependent mechanism. Parasite strains that do not induce cerebral malaria were phagocytosed and cross-presented less efficiently than PbA in vitro. The main source of antigen appears to be free merozoites, which were avidly phagocytosed. A human brain endothelial cell line also phagocytosed P. falciparum merozoites. Besides being the first demonstration of cross-presentation by brain endothelial cells, our results suggest that interfering with merozoite phagocytosis or antigen processing may be effective strategies for cerebral malaria intervention.
Multiple myeloma (MM) is an age-dependent hematological malignancy. Evaluation of immune interactions that drive MM relies on in vitro experiments that do not reflect the complex cellular stroma involved in MM pathogenesis. Here we used Vk*MYC transgenic mice, which spontaneously develop MM, and demonstrated that the immune system plays a critical role in the control of MM progression and the response to treatment. We monitored Vk*MYC mice that had been crossed with Cd226 mutant mice over a period of 3 years and found that CD226 limits spontaneous MM development. The CD226-dependent anti-myeloma immune response against transplanted Vk*MYC MM cells was mediated both by NK and CD8+ T cells through perforin and IFN-gamma pathways. Moreover, CD226 expression was required for optimal antimyeloma efficacy of cyclophosphamide (CTX) and bortezomib (Btz), which are both standardly used to manage MM in patients. Activation of costimulatory receptor CD137 with mAb (4-1BB) exerted strong antimyeloma activity, while inhibition of coinhibitory receptors PD-1 and CTLA-4 had no effect. Taken together, the results of this study provide in vivo evidence that CD226 is important for MM immunosurveillance and indicate that specific immune components should be targeted for optimal MM treatment efficacy. As progressive immunosuppression associates with MM development, strategies aimed to increase immune functions may have important therapeutic implications in MM.
Harnessing the immune adjuvant properties of natural killer T (NKT) cells is an effective strategy to generate anticancer immunity. The objective of this study was to increase the potency and durability of vaccine-induced immunity against B cell lymphoma by combining alpha-galactosylceramide (alpha-GalCer)-loaded tumor cell vaccination with an agonistic antibody targeting the immune checkpoint molecule 4-1BB (CD137). We observed potent synergy when combining vaccination and anti-4-1BB antibody treatment resulting in significantly enhanced survival of mice harboring Emu-myc tumors, including complete eradication of lymphoma in over 50% of mice. Tumor-free survival required interferon gamma (IFNgamma)-dependent expansion of CD8+ T cells and was associated with 4-1BB-mediated differentiation of KLRG1+ effector CD8+ T cells. ‘Cured’ mice were also resistant to lymphoma re-challenge 80 days later indicating successful generation of immunological memory. Overall, our results demonstrate that therapeutic anticancer vaccination against B cell lymphoma using an NKT cell ligand can be boosted by subsequent co-stimulation through 4-1BB leading to a sustainable immune response that may enhance outcomes to conventional treatment.
PURPOSE: Monoclonal antibodies (mAb) that block programmed death (PD)-1 or cytotoxic T lymphocyte antigen (CTLA-4) receptors have been associated with durable clinical responses against a variety of cancer types and hold great potential as novel cancer therapeutics. Recent evidence suggest that targeted blockade of multiple immunosuppressive pathways can induce synergistic antitumor responses. EXPERIMENTAL DESIGN: In this study, we investigated whether targeted blockade of CD73, an ectonucleotidase that catabolizes the hydrolysis of extracellular adenosine monophosphate (AMP) to adenosine, can enhance the antitumor activity of anti-CTLA-4 and anti-PD-1 mAbs against transplanted and chemically induced mouse tumors. RESULTS: Anti-CD73 mAb significantly enhanced the activity of both anti-CTLA-4 and anti-PD-1 mAbs against MC38-OVA (colon) and RM-1 (prostate) subcutaneous tumors, and established metastatic 4T1.2 breast cancer. Anti-CD73 mAb also significantly enhanced the activity of anti-PD-1 mAb against 3-methylcholanthrene (MCA)-induced fibrosarcomas. Gene-targeted mice revealed that single-agent therapies and combinatorial treatments were dependent on host IFN-gamma and CD8(+) T cells, but independent of perforin. Interestingly, anti-CD73 mAb preferentially synergized with anti-PD-1 mAb. We investigated the effect of extracellular adenosine on tumor-infiltrating T cells and showed that activation of A2A adenosine receptor enhances PD-1 expression, but not CTLA-4 expression, on tumor-specific CD8+ T cells and CD4+ Foxp3+ T regulatory cells. CONCLUSIONS: Taken together, our study revealed that targeted blockade of CD73 can enhance the therapeutic activity of anti-PD-1 and anti-CTLA-4 mAbs and may thus potentiate therapeutic strategies targeting immune checkpoint inhibitors in general.
CD4(+) T cell help contributes critically to DC-induced CD8(+) CTL immunity. However, precisely how these three cell populations interact and how CD4(+) T cell signals are delivered to CD8(+) T cells in vivo have been unclear. In this study, we developed a novel, two-step approach, wherein CD4(+) T cells and antigen-presenting DCs productively engaged one another in vivo in the absence of cognate CD8(+) T cells, after which, we selectively depleted the previously engaged CD4(+) T cells or DCs before allowing interactions of either population alone with naive CD8(+) T cells. This protocol thus allows us to clearly document the importance of CD4(+) T-licensed DCs and DC-primed CD4(+) T cells in CTL immunity. Here, we provide direct in vivo evidence that primed CD4(+) T cells or licensed DCs can stimulate CTL response and memory, independent of DC-CD4(+) T cell clusters. Our results suggest that primed CD4(+) T cells with acquired pMHC-I from DCs represent crucial “immune intermediates” for rapid induction of CTL responses and for functional memory via CD40L signaling. Importantly, intravital, two-photon microscopy elegantly provide unequivocal in vivo evidence for direct CD4-CD8(+) T cell interactions via pMHC-I engagement. This study corroborates the coexistence of direct and indirect mechanisms of T cell help for a CTL response in noninflammatory situations. These data suggest a new “dynamic model of three-cell interactions” for CTL immunity derived from stimulation by dissociated, licensed DCs, primed CD4(+) T cells, and DC-CD4(+) T cell clusters and may have significant implications for autoimmunity and vaccine design.
It is becoming increasingly evident that radiotherapy may benefit from coincident or subsequent immunotherapy. In this study, we examined whether the antitumor effects of radiotherapy, in established triple-negative breast tumors could be enhanced with combinations of clinically relevant monoclonal antibodies (mAb), designed to stimulate immunity [anti-(alpha)-CD137, alpha-CD40] or relieve immunosuppression [alpha-programmed death (PD)-1]. While the concomitant targeting of the costimulatory molecules CD137 and CD40 enhanced the antitumor effects of radiotherapy and promoted the rejection of subcutaneous BALB/c-derived 4T1.2 tumors, this novel combination was noncurative in mice bearing established C57BL/6-derived AT-3 tumors. We identified PD-1 signaling within the AT-3 tumors as a critical limiting factor to the therapeutic efficacy of alpha-CD137 therapy, alone and in combination with radiotherapy. Strikingly, all mice bearing established orthotopic AT-3 mammary tumors were cured when alpha-CD137 and alpha-PD-1 mAbs were combined with single- or low-dose fractionated radiotherapy. CD8+ T cells were essential for curative responses to this combinatorial regime. Interestingly, CD137 expression on tumor-associated CD8+ T cells was largely restricted to a subset that highly expressed PD-1. These CD137+PD-1High CD8+ T cells, persisted in irradiated AT-3 tumors, expressed Tim-3, granzyme B and Ki67 and produced IFN-gamma ex vivo in response to phorbol 12-myristate 13-acetate (PMA) and ionomycin stimulation. Notably, radiotherapy did not deplete, but enriched tumors of functionally active, tumor-specific effector cells. Collectively, these data show that concomitant targeting of immunostimulatory and inhibitory checkpoints with immunomodulatory mAbs can enhance the curative capacity of radiotherapy in established breast malignancy.
Previous studies have suggested that naive CD8 T cells require self-peptide-major histocompatability complex (MHC) complexes for maintenance. However, interpretation of such studies is complicated because of the involvement of lymphopenic animals, as lymphopenia drastically alters naive T cell homeostasis and function. In this study, we explored naive CD8 T cell survival and function in nonlymphopenic conditions by using bone marrow chimeric donors and hosts in which class I MHC expression is absent or limited to radiosensitive versus radioresistant cells. We found that long-term survival of naive CD8 T cells (but not CD4 T cells) was impaired in the absence of class I MHC. However, distinct from this effect, class I MHC deprivation also enhanced naive CD8 T cell responsiveness to low-affinity (but not high-affinity) peptide-MHC ligands. We found that this improved sensitivity was a consequence of up-regulated CD8 levels, which was mediated through a transcriptional mechanism. Hence, our data suggest that, in a nonlymphopenic setting, self-class I MHC molecules support CD8 T cell survival, but that these interactions also attenuate naive T cell sensitivity by dynamic tuning of CD8 levels.
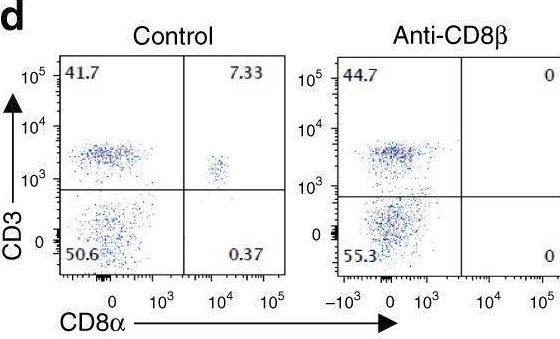
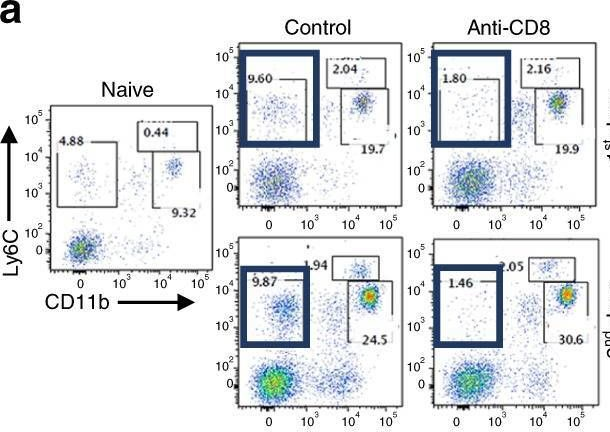
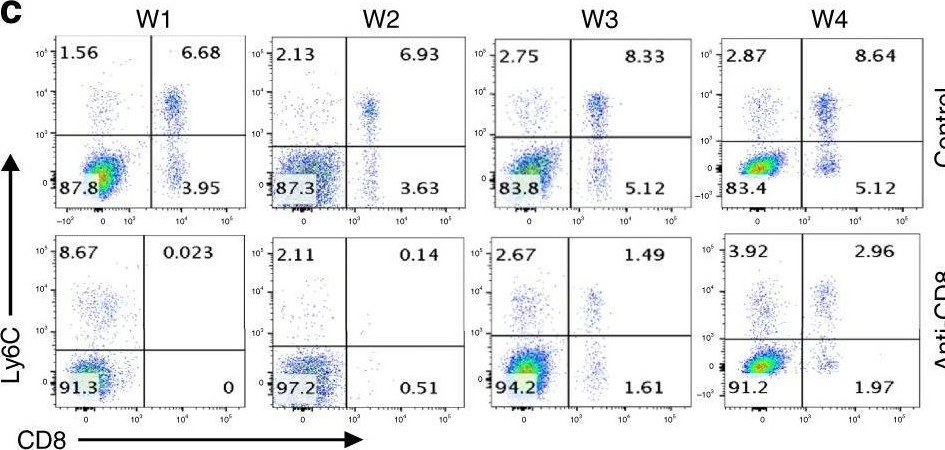
In Nature Communications on 2 April 2025 by Tsao, L. C., Wang, J. S., et al.
Trastuzumab deruxtecan (T-DXd) is an antibody-drug conjugate (ADC) targeting HER2, exhibiting significant clinical efficacy in breast cancer (BC) with varying HER2 expression, including HER2-low and HER2-ultralow. However, the precise mechanism underlying its efficacy and the contribution of immune activation in these settings remain unclear. Here, we demonstrate that T-DXd efficacy in HER2-low and HER2-negative BC is independent of HER2 engagement and ADC internalization. Instead, its activity relies on extracellular proteases, such as cathepsin L (CTSL), within the tumor microenvironment. Irrespective of their HER2 status, tumor and stromal compartments of invasive BC abundantly express CTSL, which efficiently cleaves the specialized linker of T-DXd, facilitating payload release and inducing cytotoxicity against HER2-low/negative tumors. In HER2-positive BC, the antibody backbone of T-DXd engages Fcγ-receptors and drives antibody-dependent cellular phagocytosis (ADCP). Concurrently, its cytotoxic payload (DXd) induces immunogenic cell death, further activating myeloid cells via TLR4 and STING pathways to enhance tumor antigen presentation to CD8+ T cells. Notably, T-DXd cytotoxicity also upregulates tumor CD47 expression, dampening immune activation. Combining T-DXd with CD47 checkpoint blockade significantly enhances anti-tumor immune responses in a HER2-transgenic BC mouse model, while also inducing durable CD8+ T cell memory to prevent tumor recurrence after therapy cessation. © 2025. The Author(s).
Preprint on BioRxiv : the Preprint Server for Biology on 16 February 2025 by Chi, F., Zhang, Q., et al.
A critical question in physiology is understanding how tissues adapt and alter their cellular composition in response to dietary cues. The mammalian small intestine, a vital digestive organ that absorbs nutrients, is maintained by rapidly renewing Lgr5 + intestinal stem cells (ISCs) at the intestinal crypt base. While Lgr5 + ISCs drive intestinal adaptation by altering self-renewal and differentiation divisions in response to diverse diets such as high-fat diets and fasting regimens, little is known about how micronutrients, particularly amino acids, instruct Lgr5 + ISC fate decisions to control intestinal homeostasis and repair after injury. Here, we demonstrate that cysteine, an essential amino acid, enhances the ability of Lgr5 + ISCs to repair intestinal injury. Mechanistically, the effects of cysteine on ISC-driven repair are mediated by elevated IL-22 from intraepithelial CD8αβ + T cells. These findings highlight how coupled cysteine metabolism between ISCs and CD8 + T cells augments intestinal stemness, providing a dietary approach that exploits ISC and immune cell crosstalk for ameliorating intestinal damage.
Preprint on BioRxiv : the Preprint Server for Biology on 2 February 2025 by Lim, B. J. W., Liu, M., et al.
ABSTRACT Neoadjuvant immunotherapy seeks to harness the primary tumor as a source of relevant tumor antigens to enhance systemic anti-tumor immunity through improved immunological surveillance. Despite having revolutionized the treatment of patients with high-risk early-stage triple-negative breast cancer (TNBC), a significant portion of patients remain unresponsive and succumb to metastatic recurrence post-treatment. Here, we found that optimally scheduled neoadjuvant administration of anti-4-1BB monotherapy was able to counteract metastases and prolong survival following surgical resection. Phenotypic and transcriptional profiling revealed enhanced 4-1BB expression on tumor-infiltrating intermediate (T int ), relative to progenitor (T prog ) and terminally exhausted (T term ) T cells. Furthermore, T int was enriched in low-affinity T cells. Treatment with anti-4-1BB drove clonal expansion of T int , with reduced expression of tissue-retention marker CD103 in T prog . This was accompanied by increased TCR clonotype sharing between paired tumors and pre-metastatic lungs. Further interrogation of sorted intratumor T cells confirmed enhanced T cell egress into circulation following anti-4-1BB treatment. In addition, gene signature extracted from anti-4-1BB treated T int was consistently associated with improved clinical outcomes in BRCA patients. Combinatorial neoadjuvant anti-4-1BB and ablation of tumor-derived CXCL16 resulted in enhanced therapeutic effect. These findings illustrate the intratumor changes underpinning the efficacy of neoadjuvant anti-4-1BB, highlighting the reciprocity between local tissue-retention and distant immunologic fortification, suggesting treatment can reverse the siphoning of intratumor T cells to primary tumor, enabling redistribution to distant tissues and subsequent protection against metastases.
In Nature Communications on 2 January 2025 by Lau, V. W. C., Mead, G. J., et al.
Loss of IFNγ-sensitivity by tumours is thought to be a mechanism enabling evasion, but recent studies suggest that IFNγ-resistant tumours can be sensitised for immunotherapy, yet the underlying mechanism remains unclear. Here, we show that IFNγ receptor-deficient B16-F10 mouse melanoma tumours are controlled as efficiently as WT tumours despite their lower MHC class I expression. Mechanistically, IFNγ receptor deletion in B16-F10 tumours increases IFNγ availability, triggering a remodelling of the immune landscape characterised by inflammatory monocyte infiltration and the generation of 'mono-macs'. This altered myeloid compartment synergises with an increase in antigen-specific CD8+ T cells to promote anti-tumour immunity against IFNγ receptor-deficient tumours, with such an immune crosstalk observed around blood vessels. Importantly, analysis of transcriptomic datasets suggests that similar immune remodelling occurs in human tumours carrying mutations in the IFNγ pathway. Our work thus serves mechanistic insight for the crosstalk between tumour IFNγ resistance and anti-tumour immunity, and implicates this regulation for future cancer therapy. © 2024. The Author(s).
In Cancer Research on 4 November 2024 by Jiao, M., Pirozzi, C. J., et al.
Radiotherapy (RT) is commonly used to try to eliminate any remaining tumor cells following surgical resection of glioma. However, tumor recurrence is prevalent, highlighting the unmet medical need to develop therapeutic strategies to enhance the efficacy of RT in glioma. Focusing on the radiosensitizing potential of the currently approved drugs known to cross the blood-brain barrier can facilitate rapid clinical translation. Here, we assessed the role of catechol-O-methyltransferase (COMT), a key enzyme to degrade catecholamines and a drug target for Parkinson's disease, in glioma treatment. Analysis of The Cancer Genome Atlas data showed significantly higher COMT expression levels in both low-grade glioma and glioblastoma compared to normal brain tissues. Inhibition of COMT by genetic knockout or FDA-approved COMT inhibitors significantly sensitized glioma cells to RT in vitro and in vivo. Mechanistically, COMT inhibition in glioma cells led to mitochondria dysfunction and increased mitochondrial RNA release into the cytoplasm, activating the cellular antiviral double-stranded RNA sensing pathway and type I interferon (IFN) response. Elevated type I IFNs stimulated the phagocytic capacity of microglial cells, enhancing RT efficacy. Given the long-established safety record of the COMT inhibitors, these findings provide a solid rationale to evaluate them in combination with RT in patients with glioma. Significance: Inhibition of catechol-O-methyltransferase, a well-established drug target in Parkinson's disease, interferes with mitochondrial electron transport and induces mitochondrial double-stranded RNA leakage, activating type I interferon signaling and sensitizing glioma to radiotherapy. ©2024 The Authors; Published by the American Association for Cancer Research.
In Cancer Immunology Research on 4 November 2024 by Georgiev, P., Han, S., et al.
Progressive decline of the adaptive immune system with increasing age coincides with a sharp increase in cancer incidence. In this study, we set out to understand whether deficits in antitumor immunity with advanced age promote tumor progression and/or drive resistance to immunotherapy. We found that multiple syngeneic cancers grew more rapidly in aged versus young adult mice, driven by dysfunctional CD8+ T-cell responses. By systematically mapping immune cell profiles within tumors, we identified loss of tumor antigen-specific CD8+ T cells as a primary feature accelerating the growth of tumors in aged mice and driving resistance to immunotherapy. When antigen-specific T cells from young adult mice were administered to aged mice, tumor outgrowth was delayed and the aged animals became sensitive to PD-1 blockade. These studies reveal how age-associated CD8+ T-cell dysfunction may license tumorigenesis in elderly patients and have important implications for the use of aged mice as preclinical models of aging and cancer. ©2024 The Authors; Published by the American Association for Cancer Research.
In Nature Communications on 3 October 2024 by Tan, L. M., Yin, T., et al.
Immunotherapy successfully complements traditional cancer treatment. However, primary and acquired resistance might limit efficacy. Reduced antigen presentation by MHC-I has been identified as potential resistance factor. Here we show that the epigenetic regulator ubiquitin-like with PHD and ring finger domains 1 (UHRF1), exhibits altered expression and aberrant cytosolic localization in cancerous tissues, where it promotes MHC-I ubiquitination and degradation. Cytoplasmic translocation of UHRF1 is induced by its phosphorylation on a specific serine in response to signals provided by factors present in the tumor microenvironment (TME), such as TGF-β, enabling UHRF1 to bind MHC-I. Downregulation of MHC-I results in suppression of the antigen presentation pathway to establish an immune hostile TME. UHRF1 inactivation by genetic deletion synergizes with immune checkpoint blockade (ICB) treatment and induces an anti-tumour memory response by evoking low-affinity T cells. Our study adds to the understanding of UHRF1 in cancer immune evasion and provides a potential target to synergize with immunotherapy and overcome immunotherapeutic resistance. © 2024. The Author(s).
In Nature Communications on 3 October 2024 by Tan, L. M., Yin, T., et al.
Immunotherapy successfully complements traditional cancer treatment. However, primary and acquired resistance might limit efficacy. Reduced antigen presentation by MHC-I has been identified as potential resistance factor. Here we show that the epigenetic regulator ubiquitin-like with PHD and ring finger domains 1 (UHRF1), exhibits altered expression and aberrant cytosolic localization in cancerous tissues, where it promotes MHC-I ubiquitination and degradation. Cytoplasmic translocation of UHRF1 is induced by its phosphorylation on a specific serine in response to signals provided by factors present in the tumor microenvironment (TME), such as TGF-β, enabling UHRF1 to bind MHC-I. Downregulation of MHC-I results in suppression of the antigen presentation pathway to establish an immune hostile TME. UHRF1 inactivation by genetic deletion synergizes with immune checkpoint blockade (ICB) treatment and induces an anti-tumour memory response by evoking low-affinity T cells. Our study adds to the understanding of UHRF1 in cancer immune evasion and provides a potential target to synergize with immunotherapy and overcome immunotherapeutic resistance. © 2024. The Author(s).
In Nature Cell Biology on 1 October 2024 by Yuzhalin, A. E., Lowery, F. J., et al.
Brain metastases (BrMs) evade the immune response to develop in the brain, yet the mechanisms of BrM immune evasion remains unclear. This study shows that brain astrocytes induce the overexpression of neuronal-specific cyclin-dependent kinase 5 (Cdk5) in breast cancer-derived BrMs, which facilitates BrM outgrowth in mice. Cdk5-overexpressing BrMs exhibit reduced expression and function of the class I major histocompatibility complex (MHC-I) and antigen-presentation pathway, which are restored by inhibiting Cdk5 genetically or pharmacologically, as evidenced by single-cell RNA sequencing and functional studies. Mechanistically, Cdk5 suppresses MHC-I expression on the cancer cell membrane through the Irf2bp1-Stat1-importin α-Nlrc5 pathway, enabling BrMs to avoid recognition by T cells. Treatment with roscovitine-a clinically applicable Cdk5 inhibitor-alone or combined with immune checkpoint inhibitors, significantly reduces BrM burden and increases tumour-infiltrating functional CD8+ lymphocytes in mice. Thus, astrocyte-induced Cdk5 overexpression endorses BrM immune evasion, whereas therapeutically targeting Cdk5 markedly improves the efficacy of immune checkpoint inhibitors and inhibits BrM growth. © 2024. The Author(s), under exclusive licence to Springer Nature Limited.
In The Journal of Clinical Investigation on 29 August 2024 by Janova, H., Zhao, F. R., et al.
Intestinal dysmotility syndromes have been epidemiologically associated with several antecedent bacterial and viral infections. To model this phenotype, we previously infected mice with the neurotropic flavivirus West Nile virus (WNV) and demonstrated intestinal transit defects. Here, we found that within 1 week of WNV infection, enteric neurons and glia became damaged, resulting in sustained reductions of neuronal cells and their networks of connecting fibers. Using cell-depleting antibodies, adoptive transfer experiments, and mice lacking specific immune cells or immune functions, we show that infiltrating WNV-specific CD4+ and CD8+ T cells damaged the enteric nervous system (ENS) and glia, which led to intestinal dysmotility; these T cells used multiple and redundant effector molecules including perforin and Fas ligand. In comparison, WNV-triggered ENS injury and intestinal dysmotility appeared to not require infiltrating monocytes, and damage may have been limited by resident muscularis macrophages. Overall, our experiments support a model in which antigen-specific T cell subsets and their effector molecules responding to WNV infection direct immune pathology against enteric neurons and supporting glia that results in intestinal dysmotility.
In PLoS ONE on 6 August 2024 by Morihara, H., Yamada, T., et al.
Inhibiting the cytotoxic T-lymphocyte-associated protein-4 (CTLA-4)-mediated immune checkpoint system using an anti-CTLA-4 antibody (Ab) can suppress the growth of various cancers, but the detailed mechanisms are unclear. In this study, we established a monoclonal hepatocellular carcinoma cell line (Hepa1-6 #12) and analyzed the mechanisms associated with anti-CTLA-4 Ab treatment. Depletion of CD4+ T cells, but not CD8+ T cells, prevented anti-CTLA-4 Ab-mediated anti-tumor effects, suggesting dependence on CD4+ T cells. Anti-CTLA-4 Ab treatment resulted in recruitment of interferon-gamma (IFN-g)-producing CD4+ T cells, called T-helper 1 (Th1), into tumors, and neutralization of IFN-g abrogated the anti-tumor effects. Moreover, tumor growth suppression did not require major histocompatibility complex (MHC)-I or MHC-II expression on cancer cells. In vitro studies showed that IFN-g can induce cell cycle arrest and apoptosis in tumor cells. Taken together, these data demonstrate that anti-CTLA-4 Ab can exert its anti-tumor effects through Th1-mediated cell cycle arrest and apoptosis. Copyright: © 2024 Morihara et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
In IScience on 19 July 2024 by Jiao, M., Hu, M., et al.
von Hippel-Lindau (VHL), known as a tumor suppressor gene, is frequently mutated in clear cell renal cell carcinoma (ccRCC). However, VHL mutation is not sufficient to promote tumor formation. In most cases other than ccRCC, VHL loss alters cellular homeostasis and causes cell stress and metabolic changes by stabilizing hypoxia-inducible factor (HIF) levels, resulting in a fitness disadvantage. In addition, the function of VHL in regulating immune response is still not well established. In this study, we demonstrate that VHL loss enhances the efficacy of anti-programmed death 1 (PD1) treatment in multiple murine tumor models in a T cell-dependent manner. Mechanistically, we discovered that upregulation of HIF1α/2α induced by VHL loss decreased mitochondrial outer membrane potential and caused the cytoplasmic leakage of mitochondrial DNA, which triggered cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING) activation and induced type I interferons. Our study thus provided mechanistic insights into the role of VHL gene loss in boosting antitumor immunity.© 2024 The Authors.
In Cancer Immunology, Immunotherapy : CII on 2 July 2024 by Liu, S., Wang, P., et al.
Tissue-resident memory CD103+CD8+ T cells (CD103+CD8+ TRMs) are important components of anti-tumor immunity. However, the significance of CD103+CD8+ TRMs in colorectal cancer (CRC) and their advantages remain unclear. Clinical data and specimens were used to evaluate the significance of CD103+CD8+ TRMs in CRC. A mouse subcutaneous tumorigenesis model and colony-formation assay were conducted to evaluate the anti-tumor effects of CD103+CD8+ TRMs. Finally, the infiltration density and function of CD103+CD8+ TRMs in the tumors were evaluated using flow cytometry. In this study, we showed that highly infiltrated CD103+CD8+ TRMs were associated with earlier clinical stage and negative VEGF expression in CRC patients and predicted a favorable prognosis for CRC/CRC liver metastases patients. Interestingly, we also found that CD103+CD8+ TRMs may have predictive potential for whether CRC develops liver metastasis in CRC. In addition, we found a positive correlation between the ratio of the number of α-SMA+ vessels to the sum of the number of α-SMA+ and CD31+ vessels in CRC, and the infiltration level of CD103+CD8+ TRMs. In addition, anti-angiogenic therapy promoted infiltration of CD103+CD8+ TRMs and enhanced their ability to secrete interferon (IFN)-γ, thus further improving the anti-tumor effect. Moreover, in vivo experiments showed that compared with peripheral blood CD8+ T cells, CD103+CD8+ TRMs infused back into the body could also further promote CD8+ T cells to infiltrate the tumor, and they had a stronger ability to secrete IFN-γ, which resulted in better anti-tumor effects. We demonstrated that CD103+CD8+ TRMs have the potential for clinical applications and provide new ideas for combined anti-tumor therapeutic strategies, such as anti-tumor angiogenesis therapy and CAR-T combined immunotherapy. © 2024. The Author(s).
In Cancer Immunology Research on 2 July 2024 by Praharaj, M., Shen, F., et al.
Glutamine metabolism in tumor microenvironments critically regulates antitumor immunity. Using the glutamine-antagonist prodrug JHU083, we report potent tumor growth inhibition in urologic tumors by JHU083-reprogrammed tumor-associated macrophages (TAMs) and tumor-infiltrating monocytes. We show JHU083-mediated glutamine antagonism in tumor microenvironments induced by TNF, proinflammatory, and mTORC1 signaling in intratumoral TAM clusters. JHU083-reprogrammed TAMs also exhibited increased tumor cell phagocytosis and diminished proangiogenic capacities. In vivo inhibition of TAM glutamine consumption resulted in increased glycolysis, a broken tricarboxylic acid (TCA) cycle, and purine metabolism disruption. Although the antitumor effect of glutamine antagonism on tumor-infiltrating T cells was moderate, JHU083 promoted a stem cell-like phenotype in CD8+ T cells and decreased the abundance of regulatory T cells. Finally, JHU083 caused a global shutdown in glutamine-utilizing metabolic pathways in tumor cells, leading to reduced HIF-1α, c-MYC phosphorylation, and induction of tumor cell apoptosis, all key antitumor features. Altogether, our findings demonstrate that targeting glutamine with JHU083 led to suppressed tumor growth as well as reprogramming of immunosuppressive TAMs within prostate and bladder tumors that promoted antitumor immune responses. JHU083 can offer an effective therapeutic benefit for tumor types that are enriched in immunosuppressive TAMs. ©2024 The Authors; Published by the American Association for Cancer Research.
In Cancer Immunology Research on 2 July 2024 by Praharaj, M., Shen, F., et al.
Glutamine metabolism in tumor microenvironments critically regulates antitumor immunity. Using the glutamine-antagonist prodrug JHU083, we report potent tumor growth inhibition in urologic tumors by JHU083-reprogrammed tumor-associated macrophages (TAMs) and tumor-infiltrating monocytes. We show JHU083-mediated glutamine antagonism in tumor microenvironments induced by TNF, proinflammatory, and mTORC1 signaling in intratumoral TAM clusters. JHU083-reprogrammed TAMs also exhibited increased tumor cell phagocytosis and diminished proangiogenic capacities. In vivo inhibition of TAM glutamine consumption resulted in increased glycolysis, a broken tricarboxylic acid (TCA) cycle, and purine metabolism disruption. Although the antitumor effect of glutamine antagonism on tumor-infiltrating T cells was moderate, JHU083 promoted a stem cell-like phenotype in CD8+ T cells and decreased the abundance of regulatory T cells. Finally, JHU083 caused a global shutdown in glutamine-utilizing metabolic pathways in tumor cells, leading to reduced HIF-1α, c-MYC phosphorylation, and induction of tumor cell apoptosis, all key antitumor features. Altogether, our findings demonstrate that targeting glutamine with JHU083 led to suppressed tumor growth as well as reprogramming of immunosuppressive TAMs within prostate and bladder tumors that promoted antitumor immune responses. JHU083 can offer an effective therapeutic benefit for tumor types that are enriched in immunosuppressive TAMs. ©2024 The Authors; Published by the American Association for Cancer Research.
In The Journal of Experimental Medicine on 1 July 2024 by MacLean, A. J., Bonifacio, J. P., et al.
During secondary infection with influenza virus, plasma cells (PCs) develop within the lung, providing a local source of antibodies. However, the site and mechanisms that regulate this process are poorly defined. Here, we show that while circulating memory B cells entered the lung during rechallenge and were activated within inducible bronchus-associated lymphoid tissues (iBALTs), resident memory B (BRM) cells responded earlier, and their activation occurred in a different niche: directly near infected alveoli. This process required NK cells but was largely independent of CD4 and CD8 T cells. Innate stimuli induced by virus-like particles containing ssRNA triggered BRM cell differentiation in the absence of cognate antigen, suggesting a low threshold of activation. In contrast, expansion of PCs in iBALTs took longer to develop and was critically dependent on CD4 T cells. Our work demonstrates that spatially distinct mechanisms evolved to support pulmonary secondary PC responses, and it reveals a specialized function for BRM cells as guardians of the alveoli. © 2024 MacLean et al.
In Cell Reports Medicine on 18 June 2024 by Guo, H. Z., Feng, R. X., et al.
Environmental lipids are essential for fueling tumor energetics, but whether these exogenous lipids transported into cancer cells facilitate immune escape remains unclear. Here, we find that CD36, a transporter for exogenous lipids, promotes acute myeloid leukemia (AML) immune evasion. We show that, separately from its established role in lipid oxidation, CD36 on AML cells senses oxidized low-density lipoprotein (OxLDL) to prime the TLR4-LYN-MYD88-nuclear factor κB (NF-κB) pathway, and exogenous palmitate transfer via CD36 further potentiates this innate immune pathway by supporting ZDHHC6-mediated MYD88 palmitoylation. Subsequently, NF-κB drives the expression of immunosuppressive genes that inhibit anti-tumor T cell responses. Notably, high-fat-diet or hypomethylating agent decitabine treatment boosts the immunosuppressive potential of AML cells by hijacking CD36-dependent innate immune signaling, leading to a dampened therapeutic effect. This work is of translational interest because lipid restriction by US Food and Drug Administration (FDA)-approved lipid-lowering statin drugs improves the efficacy of decitabine therapy by weakening leukemic CD36-mediated immunosuppression. Copyright © 2024 The Authors. Published by Elsevier Inc. All rights reserved.
In IScience on 19 April 2024 by Josi, R., Speiser, D. E., et al.
The global incidence of human papillomavirus (HPV) associated head and neck carcinoma is on the rise, in response to this a tetravalent therapeutic vaccine named Qβ-HPVag was developed. This vaccine, utilizing virus-like particles (VLPs) loaded with toll-like receptor ligands and chemically coupled to four HPV16-derived peptides, demonstrated strong anti-tumor effects in a murine head and neck cancer model. Qβ-HPVag impeded tumor progression, increased infiltration of HPV-specific T cells, and significantly improved survival. The vaccine`s efficacy was associated with immune repolarization in the tumor microenvironment, characterized by expanded activated dendritic cell subsets (cDC1, cDC2, DC3). Notably, mice responding to treatment exhibited a higher percentage of migratory DC3 cells expressing CCR7. These findings suggest promising prospects for optimized VLP-based vaccines in treating HPV-associated head and neck cancer. © 2024 The Author(s).
In Cancer Immunology, Immunotherapy : CII on 30 March 2024 by Xue, H., Zhang, Z., et al.
Poliovirus receptor-related immunoglobulin domain-containing protein, or PVRIG, is a newly discovered immune checkpoint that has emerged as a promising target for cancer immunotherapy. It is primarily expressed on activated T and natural killer (NK) cells, and once engaged with its ligand, PVRL2, it induces inhibitory signaling in T cells, thereby promoting the functional exhaustion of tumor-infiltrating lymphocytes (TILs). Here, we characterized IBI352g4a, a novel humanized anti-PVRIG antibody with Fc-competent function, explored the mechanism of its antitumor activity in preclinical models, and systemically evaluated the contribution of FcrR engagement to PVRIG blockade-induced antitumor activity. IBI352g4a binds to the extracellular domain of human PVRIG with high affinity (Kd = 0.53 nM) and specificity, and fully blocks the interaction between PVRIG and its ligand PVRL2. Unlike other immune checkpoints, IBI352g4a significantly induced NK cell activation and degranulation, but had a minimal effect on T-cell activation in in vitro functional assays. IBI352g4a induced strong antitumor effect in several preclinic models, through in vivo mechanism analysis we found that both NK and T cells contribute to the antitumor effect, but NK cells play predominant roles. Specifically, a single dose of IBI352g4a induced significant NK cell activation in TILs, but T-cell activation was observed only after the second dose. Moreover, the Fc effector function is critical for both NK cell activation and treatment efficacy in vitro and in vivo. Our study, for the first time, demonstrates that both NK activation and FcrR engagement are required for antitumor efficacy induced by PVRIG blockade. © 2024. The Author(s).
Preprint on BioRxiv : the Preprint Server for Biology on 28 March 2024 by Lau, V. W., Mead, G., et al.
Loss of IFNγ-sensitivity by tumours is thought to be a mechanism enabling evasion, as some cancers lacking IFNγ-signalling demonstrate resistance to checkpoint immunotherapy. However, recent studies demonstrated that IFNγ-resistant tumours are well-controlled and sensitized for immunotherapy. The underlying mechanism leading to enhanced immune responses in those patients is unknown. Using IFNγ-insensitive melanoma tumours which were well-controlled by the endogenous anti-tumour response, we found that despite low basal MHC class I expression by tumours, CD8 + T cell infiltration was not hindered and, unexpectedly, their production of IFNγ was still important for tumour control. Mechanistically, IFNγ triggers pro-inflammatory remodelling of IFNγ-insensitive tumours, affecting the differentiation of myeloid cells. Predominantly, immunosuppressive macrophages are inhibited, while inflammatory phenotypes of monocytes and ‘mono-macs’ are preserved in IFNγ-insensitive tumours. This is supported by a co-dependency between CD8 + T cells and monocyte/macrophages, as depletion of one resulted in loss of the other. Our work demonstrates an important mechanistic understanding of how IFNγ resistance does not preclude failure of anti-tumour responses. Importantly, immune remodelling appears to be dominant in IFNγ-sensitive and IFNγ-insensitive mixed tumours, and is enriched in humans with tumours mutated in the IFNγ pathway, suggesting this may be leveraged for therapy in the future.
Notifications