InVivoMAb anti-mouse CTLA-4 (CD152)
Product Details
The 9D9 monoclonal antibody reacts with mouse CTLA-4 (cytotoxic T lymphocyte antigen-4) also known as CD152. CTLA-4 is a 33 kDa cell surface receptor encoded by the Ctla4 gene that belongs to the CD28 family of the Ig superfamily. CTLA-4 is expressed on activated T and B lymphocytes. CTLA-4 is structurally similar to the T-cell co-stimulatory protein, CD28, and both molecules bind to the B7 family members B7-1 (CD80) and B7-2 (CD86). Upon ligand binding, CTLA-4 negatively regulates cell-mediated immune responses. CTLA-4 plays roles in induction and/or maintenance of immunological tolerance, thymocyte development, and regulation of protective immunity. The critical role of CTLA-4 in immune down-regulation has been demonstrated in CTLA-4 deficient mice, which succumb at 3-5 weeks of age due to the development of a lymphoproliferative disease. CTLA-4 is among a group of inhibitory receptors being explored as cancer treatment targets through immune checkpoint blockade. This CTLA-4 antibody has been extensively used in blocking/neutralization experiments and findings from a range of tumor models have established that the in vivo treatment of 9D9 monoclonal antibody results in intra-tumoral regulatory T cell depletion.Specifications
| Isotype | Mouse IgG2b |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb mouse IgG2b isotype control, unknown specificity |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Not available or unknown |
| Reported Applications |
in vivo CTLA-4 neutralization Western blot in vivo intra-tumoral regulatory T cell depletion |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
<2EU/mg (<0.002EU/μg) Determined by LAL gel clotting assay |
| Purity |
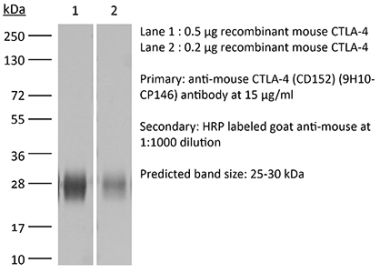
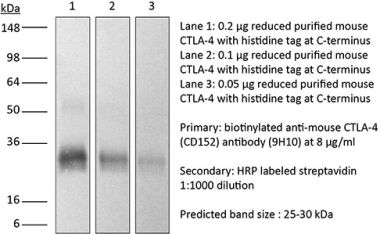
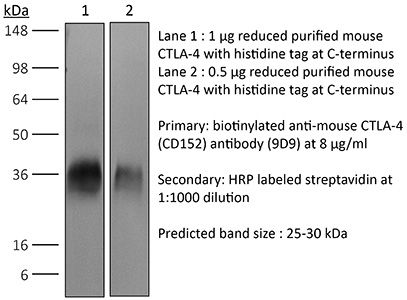
>95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein A |
| RRID | AB_10949609 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
Additional Formats




Recommended Products
-

Recommended Isotype Control(s)
InVivoMAb mouse IgG2b isotype control, unknown specificity
-

Recommended Dilution Buffer
InVivoPure pH 7.0 Dilution Buffer
in vivo CTLA-4 neutralization
Dai, M., et al. (2015). "Curing mice with large tumors by locally delivering combinations of immunomodulatory antibodies" Clin Cancer Res 21(5): 1127-1138. PubMed
PURPOSE: Immunomodulatory mAbs can treat cancer, but cures are rare except for small tumors. Our objective was to explore whether the therapeutic window increases by combining mAbs with different modes of action and injecting them into tumors. EXPERIMENTAL DESIGN: Combinations of mAbs to CD137/PD-1/CTLA-4 or CD137/PD-1/CTLA-4/CD19 were administrated intratumorally to mice with syngeneic tumors (B16 and SW1 melanoma, TC1 lung carcinoma), including tumors with a mean surface of approximately 80 mm(2). Survival and tumor growth were assessed. Immunologic responses were evaluated using flow cytometry and qRT-PCR. RESULTS: More than 50% of tumor-bearing mice had complete regression and long-term survival after tumor injection with mAbs recognizing CD137/PD-1/CTLA-4/CD19 with similar responses in three models. Intratumoral injection was more efficacious than intraperitoneal injection in causing rejection also of untreated tumors in the same mice. The three-mAb combination could also induce regression, but was less efficacious. There were few side effects, and therapy-resistant tumors were not observed. Transplanted tumor cells rapidly caused a Th2 response with increased CD19 cells. Successful therapy shifted this response to the Th1 phenotype with decreased CD19 cells and increased numbers of long-term memory CD8 effector cells and T cells making IFNgamma and TNFalpha. CONCLUSIONS: Intratumoral injection of mAbs recognizing CD137/PD-1/CTLA-4/CD19 can eradicate established tumors and reverse a Th2 response with tumor-associated CD19 cells to Th1 immunity, whereas a combination lacking anti-CD19 is less effective. There are several human cancers for which a similar approach may provide clinical benefit.
in vivo CTLA-4 neutralization
Zippelius, A., et al. (2015). "Induced PD-L1 expression mediates acquired resistance to agonistic anti-CD40 treatment" Cancer Immunol Res 3(3): 236-244. PubMed
CD40 stimulation on antigen-presenting cells (APC) allows direct activation of CD8(+) cytotoxic T cells, independent of CD4(+) T-cell help. Agonistic anti-CD40 antibodies have been demonstrated to induce beneficial antitumor T-cell responses in mouse models of cancer and early clinical trials. We report here that anti-CD40 treatment induces programmed death ligand-1 (PD-L1) upregulation on tumor-infiltrating monocytes and macrophages, which was strictly dependent on T cells and IFNgamma. PD-L1 expression could be counteracted by coadministration of antibodies blocking the PD-1 (programmed death-1)/PD-L1 axis as shown for T cells from tumor models and human donors. The combined treatment was highly synergistic and induced complete tumor rejection in about 50% of mice bearing MC-38 colon and EMT-6 breast tumors. Mechanistically, this was reflected by a strong increase of IFNgamma and granzyme-B production in intratumoral CD8(+) T cells. Concomitant CTLA-4 blockade further improved rejection of established tumors in mice. This study uncovers a novel mechanism of acquired resistance upon agonistic CD40 stimulation and proposes that the concomitant blockade of the PD-1/PD-L1 axis is a viable therapeutic strategy to optimize clinical outcomes.
in vivo CTLA-4 neutralization
Redmond, W. L., et al. (2014). "Combined targeting of costimulatory (OX40) and coinhibitory (CTLA-4) pathways elicits potent effector T cells capable of driving robust antitumor immunity" Cancer Immunol Res 2(2): 142-153. PubMed
Ligation of the TNF receptor family costimulatory molecule OX40 (CD134) with an agonist anti-OX40 monoclonal antibody (mAb) enhances antitumor immunity by augmenting T-cell differentiation as well as turning off the suppressive activity of the FoxP3(+)CD4(+) regulatory T cells (Treg). In addition, antibody-mediated blockade of the checkpoint inhibitor CTLA-4 releases the “brakes” on T cells to augment tumor immunotherapy. However, monotherapy with these agents has limited therapeutic benefit particularly against poorly immunogenic murine tumors. Therefore, we examined whether the administration of agonist anti-OX40 therapy in the presence of CTLA-4 blockade would enhance tumor immunotherapy. Combined anti-OX40/anti-CTLA-4 immunotherapy significantly enhanced tumor regression and the survival of tumor-bearing hosts in a CD4 and CD8 T cell-dependent manner. Mechanistic studies revealed that the combination immunotherapy directed the expansion of effector T-bet(high)/Eomes(high) granzyme B(+) CD8 T cells. Dual immunotherapy also induced distinct populations of Th1 [interleukin (IL)-2, IFN-gamma], and, surprisingly, Th2 (IL-4, IL-5, and IL-13) CD4 T cells exhibiting increased T-bet and Gata-3 expression. Furthermore, IL-4 blockade inhibited the Th2 response, while maintaining the Th1 CD4 and effector CD8 T cells that enhanced tumor-free survival. These data demonstrate that refining the global T-cell response during combination immunotherapy can further enhance the therapeutic efficacy of these agents.
in vivo CTLA-4 neutralization
Condamine, T., et al. (2014). "ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis" J Clin Invest 124(6): 2626-2639. PubMed
Myeloid-derived suppressor cells (MDSCs) dampen the immune response thorough inhibition of T cell activation and proliferation and often are expanded in pathological conditions. Here, we studied the fate of MDSCs in cancer. Unexpectedly, MDSCs had lower viability and a shorter half-life in tumor-bearing mice compared with neutrophils and monocytes. The reduction of MDSC viability was due to increased apoptosis, which was mediated by increased expression of TNF-related apoptosis-induced ligand receptors (TRAIL-Rs) in these cells. Targeting TRAIL-Rs in naive mice did not affect myeloid cell populations, but it dramatically reduced the presence of MDSCs and improved immune responses in tumor-bearing mice. Treatment of myeloid cells with proinflammatory cytokines did not affect TRAIL-R expression; however, induction of ER stress in myeloid cells recapitulated changes in TRAIL-R expression observed in tumor-bearing hosts. The ER stress response was detected in MDSCs isolated from cancer patients and tumor-bearing mice, but not in control neutrophils or monocytes, and blockade of ER stress abrogated tumor-associated changes in TRAIL-Rs. Together, these data indicate that MDSC pathophysiology is linked to ER stress, which shortens the lifespan of these cells in the periphery and promotes expansion in BM. Furthermore, TRAIL-Rs can be considered as potential targets for selectively inhibiting MDSCs.
in vivo CTLA-4 neutralization
Muller, P., et al. (2014). "Microtubule-depolymerizing agents used in antibody-drug conjugates induce antitumor immunity by stimulation of dendritic cells" Cancer Immunol Res 2(8): 741-755. PubMed
Antibody-drug conjugates (ADC) are emerging as powerful treatment strategies with outstanding target-specificity and high therapeutic activity in patients with cancer. Brentuximab vedotin represents a first-in-class ADC directed against CD30(+) malignancies. We hypothesized that its sustained clinical responses could be related to the stimulation of an anticancer immune response. In this study, we demonstrate that the dolastatin family of microtubule inhibitors, from which the cytotoxic component of brentuximab vedotin is derived, comprises potent inducers of phenotypic and functional dendritic cell (DC) maturation. In addition to the direct cytotoxic effect on tumor cells, dolastatins efficiently promoted antigen uptake and migration of tumor-resident DCs to the tumor-draining lymph nodes. Exposure of murine and human DCs to dolastatins significantly increased their capacity to prime T cells. Underlining the requirement of an intact host immune system for the full therapeutic benefit of dolastatins, the antitumor effect was far less pronounced in immunocompromised mice. We observed substantial therapeutic synergies when combining dolastatins with tumor antigen-specific vaccination or blockade of the PD-1-PD-L1 and CTLA-4 coinhibitory pathways. Ultimately, treatment with ADCs using dolastatins induces DC homing and activates cellular antitumor immune responses in patients. Our data reveal a novel mechanism of action for dolastatins and provide a strong rationale for clinical treatment regimens combining dolastatin-based therapies, such as brentuximab vedotin, with immune-based therapies.
in vivo CTLA-4 neutralization
Bulliard, Y., et al. (2013). "Activating Fc gamma receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies" J Exp Med 210(9): 1685-1693. PubMed
Fc gamma receptor (FcgammaR) coengagement can facilitate antibody-mediated receptor activation in target cells. In particular, agonistic antibodies that target tumor necrosis factor receptor (TNFR) family members have shown dependence on expression of the inhibitory FcgammaR, FcgammaRIIB. It remains unclear if engagement of FcgammaRIIB also extends to the activities of antibodies targeting immunoregulatory TNFRs expressed by T cells. We have explored the requirement for activating and inhibitory FcgammaRs for the antitumor effects of antibodies targeting the TNFR glucocorticoid-induced TNFR-related protein (GITR; TNFRSF18; CD357) expressed on activated and regulatory T cells (T reg cells). We found that although FcgammaRIIB was dispensable for the in vivo efficacy of anti-GITR antibodies, in contrast, activating FcgammaRs were essential. Surprisingly, the dependence on activating FcgammaRs extended to an antibody targeting the non-TNFR receptor CTLA-4 (CD152) that acts as a negative regulator of T cell immunity. We define a common mechanism that correlated with tumor efficacy, whereby antibodies that coengaged activating FcgammaRs expressed by tumor-associated leukocytes facilitated the selective elimination of intratumoral T cell populations, particularly T reg cells. These findings may have broad implications for antibody engineering efforts aimed at enhancing the therapeutic activity of immunomodulatory antibodies.
in vivo CTLA-4 neutralization
Wei, H., et al. (2013). "Combinatorial PD-1 blockade and CD137 activation has therapeutic efficacy in murine cancer models and synergizes with cisplatin" PLoS One 8(12): e84927. PubMed
90 days (and was probably curative) by a mechanism which included a systemic CD8(+) T cell response with tumor specificity and immunological memory. Strikingly, combined treatment of cisplatin and CD137/PD-1 mAb also gave rise to the long-term survival of mice with established TC1 lung tumors. A similar combination of the 2 mAbs and cisplatin should be considered for clinical ‘translation’.”}” data-sheets-userformat=”{“2″:14851,”3”:{“1″:0},”4”:{“1″:2,”2″:16777215},”12″:0,”14”:{“1″:2,”2″:1521491},”15″:”Roboto, sans-serif”,”16″:12}”>There is an urgent need for improved therapy for advanced ovarian carcinoma, which may be met by administering immune-modulatory monoclonal antibodies (mAbs) to generate a tumor-destructive immune response. Using the ID8 mouse ovarian cancer model, we investigated the therapeutic efficacy of various mAb combinations in mice with intraperitoneal (i.p.) tumor established by transplanting 3 x 10(6) ID8 cells 10 days previously. While most of the tested mAbs were ineffective when given individually or together, the data confirm our previous finding that 2 i.p. injections of a combination of anti-CD137 with anti-PD-1 mAbs doubles overall survival. Mice treated with this mAb combination have a significantly increased frequency and total number of CD8(+) T cells both in the peritoneal lavage and spleens, and these cells are functional as demonstrated by antigen-specific cytolytic activity and IFN-gamma production. While administration of anti-CD137 mAb as a single agent similarly increases CD8(+) T cells, these have no functional activity, which may be attributed to up-regulation of co-inhibitory PD-1 and TIM-3 molecules induced by CD137. Addition of the anti-cancer drug cisplatin to the 2 mAb combination increased overall survival >90 days (and was probably curative) by a mechanism which included a systemic CD8(+) T cell response with tumor specificity and immunological memory. Strikingly, combined treatment of cisplatin and CD137/PD-1 mAb also gave rise to the long-term survival of mice with established TC1 lung tumors. A similar combination of the 2 mAbs and cisplatin should be considered for clinical ‘translation’.
in vivo CTLA-4 neutralization
Dai, M., et al. (2013). "Long-lasting complete regression of established mouse tumors by counteracting Th2 inflammation" J Immunother 36(4): 248-257. PubMed
40% of mice with SW1 tumors remained healthy >150 days after last treatment and are probably cured. Therapeutic efficacy was associated with a systemic immune response with memory and antigen specificity, required CD4 cells and involved CD8 cells and NK cells to a less extent. The 3 mAb combination significantly decreased CD19 cells at tumor sites, increased IFN-gamma and TNF-alpha producing CD4 and CD8 T cells and mature CD86 dendritic cells (DC), and it increased the ratios of effector CD4 and CD8 T cells to CD4Foxp3 regulatory T (Treg) cells and to CD11bGr-1 myeloid suppressor cells (MDSC). This is consistent with shifting the tumor microenvironment from an immunosuppressive Th2 to an immunostimulatory Th1 type and is further supported by PCR data. Adding an anti-CD19 mAb to the 3 mAb combination in the SW1 model further increased therapeutic efficacy. Data from ongoing experiments show that intratumoral injection of a combination of mAbs to CD137PD-1CTLA4CD19 can induce complete regression and dramatically prolong survival also in the TC1 carcinoma and B16 melanoma models, suggesting that the approach has general validity.”}” data-sheets-userformat=”{“2″:14851,”3”:{“1″:0},”4”:{“1″:2,”2″:16777215},”12″:0,”14”:{“1″:2,”2″:1521491},”15″:”Roboto, sans-serif”,”16″:12}”>Mice with intraperitoneal ID8 ovarian carcinoma or subcutaneous SW1 melanoma were injected with monoclonal antibodies (mAbs) to CD137PD-1CTLA4 7-15 days after tumor initiation. Survival of mice with ID8 tumors tripled and >40% of mice with SW1 tumors remained healthy >150 days after last treatment and are probably cured. Therapeutic efficacy was associated with a systemic immune response with memory and antigen specificity, required CD4 cells and involved CD8 cells and NK cells to a less extent. The 3 mAb combination significantly decreased CD19 cells at tumor sites, increased IFN-gamma and TNF-alpha producing CD4 and CD8 T cells and mature CD86 dendritic cells (DC), and it increased the ratios of effector CD4 and CD8 T cells to CD4Foxp3 regulatory T (Treg) cells and to CD11bGr-1 myeloid suppressor cells (MDSC). This is consistent with shifting the tumor microenvironment from an immunosuppressive Th2 to an immunostimulatory Th1 type and is further supported by PCR data. Adding an anti-CD19 mAb to the 3 mAb combination in the SW1 model further increased therapeutic efficacy. Data from ongoing experiments show that intratumoral injection of a combination of mAbs to CD137PD-1CTLA4CD19 can induce complete regression and dramatically prolong survival also in the TC1 carcinoma and B16 melanoma models, suggesting that the approach has general validity.
in vivo CTLA-4 neutralization
Hooijkaas, A., et al. (2012). "Selective BRAF inhibition decreases tumor-resident lymphocyte frequencies in a mouse model of human melanoma" Oncoimmunology 1(5): 609-617. PubMed
The development of targeted therapies and immunotherapies has markedly advanced the treatment of metastasized melanoma. While treatment with selective BRAF(V600E) inhibitors (like vemurafenib or dabrafenib) leads to high response rates but short response duration, CTLA-4 blocking therapies induce sustained responses, but only in a limited number of patients. The combination of these diametric treatment approaches may further improve survival, but pre-clinical data concerning this approach is limited. We investigated, using Tyr::CreER(T2)PTEN(F-/-)BRAF(F-V600E/+) inducible melanoma mice, whether BRAF(V600E) inhibition can synergize with anti-CTLA-4 mAb treatment, focusing on the interaction between the BRAF(V600E) inhibitor PLX4720 and the immune system. While PLX4720 treatment strongly decreased tumor growth, it did not induce cell death in BRAF(V600E)/PTEN(-/-) melanomas. More strikingly, PLX4720 treatment led to a decreased frequency of tumor-resident T cells, NK-cells, MDSCs and macrophages, which could not be restored by the addition of anti-CTLA-4 mAb. As this effect was not observed upon treatment of BRAF wild-type B16F10 tumors, we conclude that the decreased frequency of immune cells correlates to BRAF(V600E) inhibition in tumor cells and is not due to an off-target effect of PLX4720 on immune cells. Furthermore, anti-CTLA-4 mAb treatment of inducible melanoma mice treated with PLX4720 did not result in enhanced tumor control, while anti-CTLA-4 mAb treatment did improve the effect of tumor-vaccination in B16F10-inoculated mice. Our data suggest that vemurafenib may negatively affect the immune activity within the tumor. Therefore, the potential effect of targeted therapy on the tumor-microenvironment should be taken into consideration in the design of clinical trials combining targeted and immunotherapy.
in vivo CTLA-4 neutralization
Curran, M. A., et al. (2011). "Combination CTLA-4 blockade and 4-1BB activation enhances tumor rejection by increasing T-cell infiltration, proliferation, and cytokine production" PLoS One 6(4): e19499. PubMed
BACKGROUND: The co-inhibitory receptor Cytotoxic T-Lymphocyte Antigen 4 (CTLA-4) attenuates immune responses and prevent autoimmunity, however, tumors exploit this pathway to evade the host T-cell response. The T-cell co-stimulatory receptor 4-1BB is transiently upregulated on T-cells following activation and increases their proliferation and inflammatory cytokine production when engaged. Antibodies which block CTLA-4 or which activate 4-1BB can promote the rejection of some murine tumors, but fail to cure poorly immunogenic tumors like B16 melanoma as single agents. METHODOLOGY/PRINCIPAL FINDINGS: We find that combining alphaCTLA-4 and alpha4-1BB antibodies in the context of a Flt3-ligand, but not a GM-CSF, based B16 melanoma vaccine promoted synergistic levels of tumor rejection. 4-1BB activation elicited strong infiltration of CD8+ T-cells into the tumor and drove the proliferation of these cells, while CTLA-4 blockade did the same for CD4+ effector T-cells. Anti-4-1BB also depressed regulatory T-cell infiltration of tumors. 4-1BB activation strongly stimulated inflammatory cytokine production in the vaccine and tumor draining lymph nodes and in the tumor itself. The addition of CTLA-4 blockade further increased IFN-gamma production from CD4+ effector T-cells in the vaccine draining node and the tumor. Anti 4-1BB treatment, with or without CTLA-4 blockade, induced approximately 75% of CD8+ and 45% of CD4+ effector T-cells in the tumor to express the killer cell lectin-like receptor G1 (KLRG1). Tumors treated with combination antibody therapy showed 1.7-fold greater infiltration by these KLRG1+CD4+ effector T-cells than did those treated with alpha4-1BB alone. CONCLUSIONS/SIGNIFICANCE: This study shows that combining T-cell co-inhibitory blockade with alphaCTLA-4 and active co-stimulation with alpha4-1BB promotes rejection of B16 melanoma in the context of a suitable vaccine. In addition, we identify KLRG1 as a useful marker for monitoring the anti-tumor immune response elicited by this therapy. These findings should aid in the design of future trials for the immunotherapy of melanoma.
in vivo CTLA-4 neutralization
Balachandran, V. P., et al. (2011). "Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido" Nat Med 17(9): 1094-1100. PubMed
Imatinib mesylate targets mutated KIT oncoproteins in gastrointestinal stromal tumor (GIST) and produces a clinical response in 80% of patients. The mechanism is believed to depend predominantly on the inhibition of KIT-driven signals for tumor-cell survival and proliferation. Using a mouse model of spontaneous GIST, we found that the immune system contributes substantially to the antitumor effects of imatinib. Imatinib therapy activated CD8(+) T cells and induced regulatory T cell (T(reg) cell) apoptosis within the tumor by reducing tumor-cell expression of the immunosuppressive enzyme indoleamine 2,3-dioxygenase (Ido). Concurrent immunotherapy augmented the efficacy of imatinib in mouse GIST. In freshly obtained human GIST specimens, the T cell profile correlated with imatinib sensitivity and IDO expression. Thus, T cells are crucial to the antitumor effects of imatinib in GIST, and concomitant immunotherapy may further improve outcomes in human cancers treated with targeted agents.