InVivoMAb anti-mouse PD-1 (CD279)
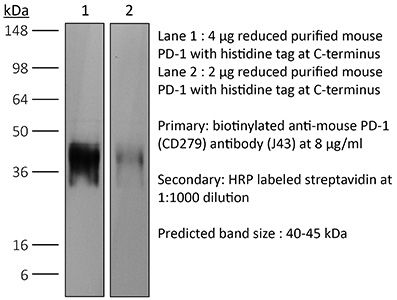
Product Details
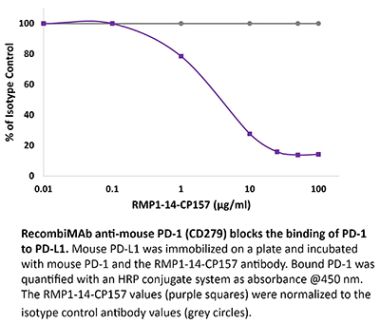
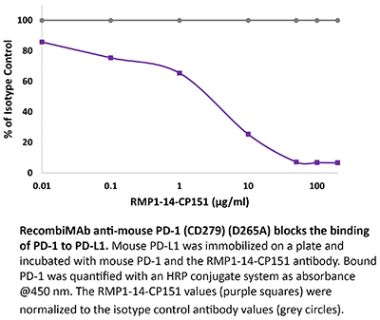
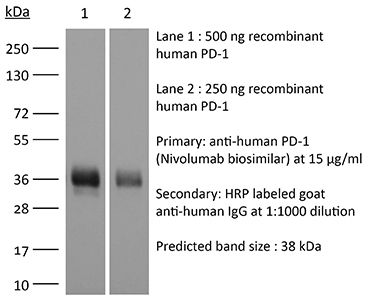
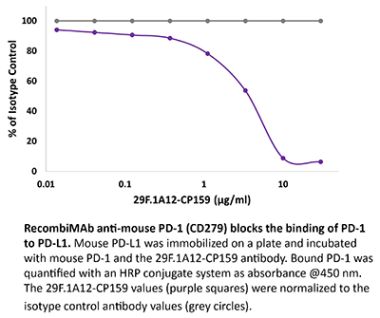
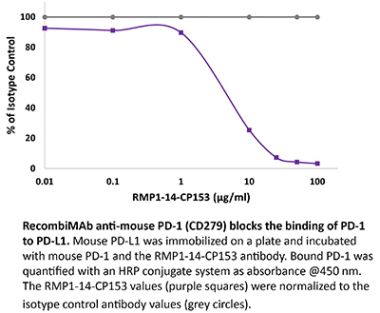
The J43 monoclonal antibody reacts with mouse PD-1 (programmed death-1) also known as CD279. PD-1 is a 50-55 kDa cell surface receptor encoded by the Pdcd1 gene that belongs to the CD28 family of the immunoglobulin superfamily. PD-1 is transiently expressed on CD4 and CD8 thymocytes as well as activated T and B lymphocytes and myeloid cells. PD-1 expression declines after successful elimination of antigen. Additionally, Pdcd1 mRNA is expressed in developing B lymphocytes during the pro-B-cell stage. PD-1’s structure includes a ITIM (immunoreceptor tyrosine-based inhibitory motif) suggesting that PD-1 negatively regulates TCR signals. PD-1 signals via binding its two ligands, PD-L1 and PD-L2 both members of the B7 family. Upon ligand binding, PD-1 signaling inhibits T-cell activation, leading to reduced proliferation, cytokine production, and T-cell death. Additionally, PD-1 is known to play key roles in peripheral tolerance and prevention of autoimmune disease in mice as PD-1 knockout animals show dilated cardiomyopathy, splenomegaly, and loss of peripheral tolerance. Induced PD-L1 expression is common in many tumors including squamous cell carcinoma, colon adenocarcinoma, and breast adenocarcinoma. PD-L1 overexpression results in increased resistance of tumor cells to CD8 T cell mediated lysis. In mouse models of melanoma, tumor growth can be transiently arrested via treatment with antibodies which block the interaction between PD-L1 and its receptor PD-1. For these reasons anti-PD-1 mediated immunotherapies are currently being explored as cancer treatments. The J43 antibody has been shown to block the binding of both mouse PD-L1-Ig and mouse PD-L2-Ig to PD-1.Specifications
| Isotype | Armenian hamster IgG |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb polyclonal Armenian hamster IgG |
| Recommended Dilution Buffer | InVivoPure pH 6.5 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Syrian Hamster BKH cells transfected with mouse PD-1 cDNA |
| Reported Applications |
in vivo blocking of PD-1/PD-L signaling in vitro PD-1 neutralization Western blot |
| Formulation |
PBS, pH 6.5 Contains no stabilizers or preservatives |
| Endotoxin |
<2EU/mg (<0.002EU/μg) Determined by LAL gel clotting assay |
| Purity |
>95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_1107747 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
Additional Formats
Recommended Products
-

Recommended Isotype Control(s)
InVivoMAb polyclonal Armenian hamster IgG
-

Recommended Dilution Buffer
InVivoPure pH 6.5 Dilution Buffer
in vivo blocking of PD-1/PD-L signaling
Li, J., et al. (2018). "Co-inhibitory Molecule B7 Superfamily Member 1 Expressed by Tumor-Infiltrating Myeloid Cells Induces Dysfunction of Anti-tumor CD8(+) T Cells" Immunity 48(4): 773-786 e775. PubMed
The molecular mechanisms whereby CD8(+) T cells become “exhausted” in the tumor microenvironment remain unclear. Programmed death ligand-1 (PD-L1) is upregulated on tumor cells and PD-1-PD-L1 blockade has significant efficacy in human tumors; however, most patients do not respond, suggesting additional mechanisms underlying T cell exhaustion. B7 superfamily member 1 (B7S1), also called B7-H4, B7x, or VTCN1, negatively regulates T cell activation. Here we show increased B7S1 expression on myeloid cells from human hepatocellular carcinoma correlated with CD8(+) T cell dysfunction. B7S1 inhibition suppressed development of murine tumors. Putative B7S1 receptor was co-expressed with PD-1 but not T cell immunoglobulin and mucin-domain containing-3 (Tim-3) at an activated state of early tumor-infiltrating CD8(+) T cells, and B7S1 promoted T cell exhaustion, possibly through Eomes overexpression. Combinatorial blockade of B7S1 and PD-1 synergistically enhanced anti-tumor immune responses. Collectively, B7S1 initiates dysfunction of tumor-infiltrating CD8(+) T cells and may be targeted for cancer immunotherapy.
in vivo blocking of PD-1/PD-L signaling
Li, C., et al. (2015). "ADAP and SKAP55 deficiency suppresses PD-1 expression in CD8+ cytotoxic T lymphocytes for enhanced anti-tumor immunotherapy" EMBO Mol Med 7(6): 754-769. PubMed
PD-1 negatively regulates CD8(+) cytotoxic T lymphocytes (CTL) cytotoxicity and anti-tumor immunity. However, it is not fully understood how PD-1 expression on CD8(+) CTL is regulated during anti-tumor immunotherapy. In this study, we have identified that the ADAP-SKAP55 signaling module reduced CD8(+) CTL cytotoxicity and enhanced PD-1 expression in a Fyn-, Ca(2+)-, and NFATc1-dependent manner. In DC vaccine-based tumor prevention and therapeutic models, knockout of SKAP55 or ADAP showed a heightened protection from tumor formation or metastases in mice and reduced PD-1 expression in CD8(+) effector cells. Interestingly, CTLA-4 levels and the percentages of tumor infiltrating CD4(+)Foxp3(+) Tregs remained unchanged. Furthermore, adoptive transfer of SKAP55-deficient or ADAP-deficient CD8(+) CTLs significantly blocked tumor growth and increased anti-tumor immunity. Pretreatment of wild-type CD8(+) CTLs with the NFATc1 inhibitor CsA could also downregulate PD-1 expression and enhance anti-tumor therapeutic efficacy. Together, we propose that targeting the unrecognized ADAP-SKAP55-NFATc1-PD-1 pathway might increase efficacy of anti-tumor immunotherapy.
in vivo blocking of PD-1/PD-L signaling
Imai, Y., et al. (2015). "Cutting Edge: PD-1 Regulates Imiquimod-Induced Psoriasiform Dermatitis through Inhibition of IL-17A Expression by Innate gammadelta-Low T Cells" J Immunol 195(2): 421-425. PubMed
Programmed cell death 1 (PD-1) is a key regulatory molecule that has been targeted in human cancers, including melanoma. In clinical testing, Abs against PD-1 have resulted in psoriasiform dermatitis (PsD). To determine whether PD-1 regulates PsD, we compared skin responses of PD-1-deficient (PD-1KO) mice and wild-type (WT) controls in an imiquimod (IMQ)-induced murine model of psoriasis. PD-1KO mice showed severe epidermal hyperplasia, greater neutrophilic infiltration, and higher expression of Th17 cytokines (versus WT mice). IMQ exposure increased PD-1 expression by skin gammadelta-low (GDL) T cells and enhanced expression of PD-L1 by keratinocytes. Three-fold increases in the percentage of IL-17A(+) GDL T cells were observed in skin cell suspensions derived from IMQ-treated PD-1KO mice (versus WT controls), suggesting that the lack of PD-1 has a functional effect not only on alphabeta T cells, but also on GDL T cells, and that PD-1 may play a regulatory role in PsD.
in vitro PD-1 neutralization
Verhagen, J. and D. C. Wraith. (2014). "Blockade of LFA-1 augments in vitro differentiation of antigen-induced Foxp3(+) Treg cells" J Immunol Methods 414: 58-64. PubMed
Adoptive transfer of antigen-specific, in vitro-induced Foxp3(+) Treg (iTreg) cells protects against autoimmune disease. To generate antigen-specific iTreg cells at high purity, however, remains a challenge. Whereas polyclonal T cell stimulation with anti-CD3 and anti-CD28 antibody yields Foxp3(+) iTreg cells at a purity of 90-95%, antigen-induced iTreg cells typically do not exceed a purity of 65-75%, even in a TCR-transgenic model. In a similar vein to thymic Treg cell selection, iTreg cell differentiation is influenced not only by antigen recognition and the availability of TGF-beta but also by co-factors including costimulation and adhesion molecules. In this study, we demonstrate that blockade of the T cell integrin Leukocyte Function-associated Antigen-1 (LFA-1) during antigen-mediated iTreg cell differentiation augments Foxp3 induction, leading to approximately 90% purity of Foxp3(+) iTreg cells. This increased efficacy not only boosts the yield of Foxp3(+) iTreg cells, it also reduces contamination with activated effector T cells, thus improving the safety of adoptive transfer immunotherapy.
in vivo blocking of PD-1/PD-L signaling
Rabenstein, H., et al. (2014). "Differential kinetics of antigen dependency of CD4+ and CD8+ T cells" J Immunol 192(8): 3507-3517. PubMed
Ag recognition via the TCR is necessary for the expansion of specific T cells that then contribute to adaptive immunity as effector and memory cells. Because CD4+ and CD8+ T cells differ in terms of their priming APCs and MHC ligands we compared their requirements of Ag persistence during their expansion phase side by side. Proliferation and effector differentiation of TCR transgenic and polyclonal mouse T cells were thus analyzed after transient and continuous TCR signals. Following equally strong stimulation, CD4+ T cell proliferation depended on prolonged Ag presence, whereas CD8+ T cells were able to divide and differentiate into effector cells despite discontinued Ag presentation. CD4+ T cell proliferation was neither affected by Th lineage or memory differentiation nor blocked by coinhibitory signals or missing inflammatory stimuli. Continued CD8+ T cell proliferation was truly independent of self-peptide/MHC-derived signals. The subset divergence was also illustrated by surprisingly broad transcriptional differences supporting a stronger propensity of CD8+ T cells to programmed expansion. These T cell data indicate an intrinsic difference between CD4+ and CD8+ T cells regarding the processing of TCR signals for proliferation. We also found that the presentation of a MHC class II-restricted peptide is more efficiently prolonged by dendritic cell activation in vivo than a class I bound one. In summary, our data demonstrate that CD4+ T cells require continuous stimulation for clonal expansion, whereas CD8+ T cells can divide following a much shorter TCR signal.
in vivo blocking of PD-1/PD-L signaling
Sarraj, B., et al. (2014). "Impaired selectin-dependent leukocyte recruitment induces T-cell exhaustion and prevents chronic allograft vasculopathy and rejection" Proc Natl Acad Sci U S A 111(33): 12145-12150. PubMed
Selectin-selectin ligand interactions mediate the initial steps in leukocyte migration, an integral part of immune responses. Fucosyltransferase-VII (FucT-VII), encoded by Fut7, is essential for biosynthesis of selectin ligands. In an established model of cardiac allograft vasculopathy and chronic rejection, Fut7(-/-) recipients exhibited long-term graft survival with minimal vasculopathy compared with WT controls. Graft survival was associated with CD4 T-cell exhaustion in the periphery, characterized by impaired effector cytokine production, defective proliferation, increased expression of inhibitory receptors programmed death-1 (PD-1) and T cell Ig- and mucin-domain-containing molecule-3 (Tim-3), low levels of IL-7Ralpha on CD4 T cells, and reduced migration of polyfunctional CD4 memory T cells to the allograft. Blocking PD-1 triggered rejection only in Fut7(-/-) recipients, whereas depleting regulatory T cells had no effect in either Fut7(-/-) or WT recipients. Adoptive transfer experiments confirmed that this CD4 T cell-exhausted phenotype is seen primarily in Fut7(-/-) CD4 T cells. These data suggest that impaired leukocyte recruitment is a novel mechanism leading to CD4 T-cell exhaustion. Our experimental system serves as an excellent model to study CD4 T-cell exhaustion as a dominant mechanism of transplant tolerance. Further, targeting FucT-VII may serve as a promising strategy to prevent chronic allograft rejection and promote tolerance.
in vivo blocking of PD-1/PD-L signaling
Van der Jeught, K., et al. (2014). "Intratumoral administration of mRNA encoding a fusokine consisting of IFN-beta and the ectodomain of the TGF-beta receptor II potentiates antitumor immunity" Oncotarget 5(20): 10100-10113. PubMed
It is generally accepted that the success of immunotherapy depends on the presence of tumor-specific CD8(+) cytotoxic T cells and the modulation of the tumor environment. In this study, we validated mRNA encoding soluble factors as a tool to modulate the tumor microenvironment to potentiate infiltration of tumor-specific T cells. Intratumoral delivery of mRNA encoding a fusion protein consisting of interferon-beta and the ectodomain of the transforming growth factor-beta receptor II, referred to as Fbeta(2), showed therapeutic potential. The treatment efficacy was dependent on CD8(+) T cells and could be improved through blockade of PD-1/PD-L1 interactions. In vitro studies revealed that administration of Fbeta(2) to tumor cells resulted in a reduced proliferation and increased expression of MHC I but also PD-L1. Importantly, Fbeta(2) enhanced the antigen presenting capacity of dendritic cells, whilst reducing the suppressive activity of myeloid-derived suppressor cells. In conclusion, these data suggest that intratumoral delivery of mRNA encoding soluble proteins, such as Fbeta(2), can modulate the tumor microenvironment, leading to effective antitumor T cell responses, which can be further potentiated through combination therapy.
in vivo blocking of PD-1/PD-L signaling, in vitro PD-1 neutralization
Park, S. J., et al. (2014). "Negative role of inducible PD-1 on survival of activated dendritic cells" J Leukoc Biol 95(4): 621-629. PubMed
PD-1 is a well-established negative regulator of T cell responses by inhibiting proliferation and cytokine production of T cells via interaction with its ligands, B7-H1 (PD-L1) and B7-DC (PD-L2), expressed on non-T cells. Recently, PD-1 was found to be expressed in innate cells, including activated DCs, and plays roles in suppressing production of inflammatory cytokines. In this study, we demonstrate that PD-1 KO DCs exhibited prolonged longevity compared with WT DCs in the dLNs after transfer of DCs into hind footpads. Interestingly, upon LPS stimulation, WT DCs increased the expression of PD-1 and started to undergo apoptosis. DCs, in spleen of LPS-injected PD-1 KO mice, were more resistant to LPS-mediated apoptosis in vivo than WT controls. Moreover, treatment of blocking anti-PD-1 mAb during DC maturation resulted in enhanced DC survival, suggesting that PD-1:PD-L interactions are involved in DC apoptosis. As a result, PD-1-deficient DCs augmented T cell responses in terms of antigen-specific IFN-gamma production and proliferation of CD4 and CD8 T cells to a greater degree than WT DCs. Moreover, PD-1 KO DCs exhibited increased MAPK1 and CD40-CD40L signaling, suggesting a possible mechanism for enhanced DC survival in the absence of PD-1 expression. Taken together, our findings further extend the function of PD-1, which plays an important role in apoptosis of activated DCs and provides important implications for PD-1-mediated immune regulation.
in vitro PD-1 neutralization
Schwager, K., et al. (2013). "The immunocytokine L19-IL2 eradicates cancer when used in combination with CTLA-4 blockade or with L19-TNF" J Invest Dermatol 133(3): 751-758. PubMed
Systemic high-dose IL2 promotes long-term survival in a subset of metastatic melanoma patients, but this treatment is accompanied by severe toxicities. The immunocytokine L19-IL2, in which IL2 is fused to the human L19 antibody capable of selective accumulation on tumor neovasculature, has recently shown encouraging clinical activity in patients with metastatic melanoma. In this study, we have investigated the therapeutic performance of L19-IL2, administered systemically in combination with a murine anti-CTLA-4 antibody or with a second clinical-stage immunocytokine (L19-TNF) in two syngeneic immunocompetent mouse models of cancer. We observed complete tumor eradications when L19-IL2 was used in combination with CTLA-4 blockade. Interestingly, mice cured from F9 tumors developed new lesions when rechallenged with tumor cells after therapy, whereas mice cured from CT26 tumors were resistant to tumor rechallenge. Similarly, L19-IL2 induced complete remissions when administered in a single intratumoral injection in combination with L19-TNF, whereas the two components did not lead to cures when administered as single agents. These findings provide a rationale for combination trials in melanoma, as the individual therapeutic agents have been extensively studied in clinical trials, and the antigen recognized by the L19 antibody has an identical sequence in mouse and man.
in vitro PD-1 neutralization
Noval Rivas, M., et al. (2009). "Reviving function in CD4+ T cells adapted to persistent systemic antigen" J Immunol 183(7): 4284-4291. PubMed
In bone marrow-transplanted patients, chronic graft-versus-host disease is a complication that results from the persistent stimulation of recipient minor histocompatibility Ag (mHA)-specific T cells contained within the graft. In this study, we developed a mouse model where persistent stimulation of donor T cells by recipient’s mHA led to multiorgan T cell infiltration. Exposure to systemic mHA, however, deeply modified T cell function and chronically stimulated T cells developed a long-lasting state of unresponsiveness, or immune adaptation, characterized by their inability to mediate organ immune damages in vivo. However, analysis of the gene expression profile of adapted CD4+ T cells revealed the specific coexpression of genes known to promote differentiation and function of Th1 effector cells as well as genes coding for proteins that control T cell activity, such as cell surface-negative costimulatory molecules and regulatory cytokines. Strikingly, blockade of negative costimulation abolished T cell adaptation and stimulated strong IFN-gamma production and severe multiorgan wasting disease. Negative costimulation was also shown to control lethal LPS-induced toxic shock in mice with adapted T cells, as well as the capacity of adapted T cells to reject skin graft. Our results demonstrate that negative costimulation is the molecular mechanism used by CD4+ T cells to adapt their activity in response to persistent antigenic stimulation. The effector function of CD4+ T cells that have adapted to chronic Ag presentation can be activated by stimuli strong enough to overcome regulatory signals delivered to the T cells by negative costimulation.