InVivoPlus anti-mouse PD-1 (CD279)

Product Details
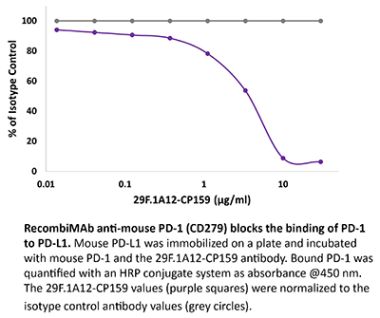
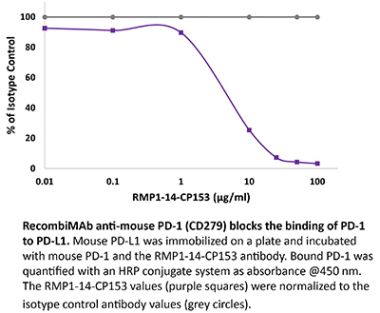
The 29F.1A12™ monoclonal antibody reacts with mouse PD-1 (programmed death-1) also known as CD279. PD-1 is a 50-55 kDa cell surface receptor encoded by the Pdcd1 gene that belongs to the CD28 family of the Ig superfamily. PD-1 is transiently expressed on CD4 and CD8 thymocytes as well as activated T and B lymphocytes and myeloid cells. PD-1 expression declines after successful elimination of antigen. Additionally, Pdcd1 mRNA is expressed in developing B lymphocytes during the pro-B-cell stage. PD-1’s structure includes a ITIM (immunoreceptor tyrosine-based inhibitory motif) suggesting that PD-1 negatively regulates TCR signals. PD-1 signals via binding its two ligands, PD-L1 and PD-L2 both members of the B7 family. Upon ligand binding, PD-1 signaling inhibits T-cell activation, leading to reduced proliferation, cytokine production, and T-cell death. Additionally, PD-1 is known to play key roles in peripheral tolerance and prevention of autoimmune disease in mice as PD-1 knockout animals show dilated cardiomyopathy, splenomegaly, and loss of peripheral tolerance. Induced PD-L1 expression is common in many tumors including squamous cell carcinoma, colon adenocarcinoma, and breast adenocarcinoma. PD-L1 overexpression results in increased resistance of tumor cells to CD8 T cell mediated lysis. In mouse models of melanoma, tumor growth can be transiently arrested via treatment with antibodies which block the interaction between PD-L1 and its receptor PD-1. For these reasons anti-PD-1 mediated immunotherapies are currently being explored as cancer treatments. Like the RMP1-14 and J43 antibodies the 29F.1A12™ antibody has been shown to block the binding of PD-1 to its ligands in vivo.Specifications
| Isotype | Rat IgG2a |
|---|---|
| Recommended Isotype Control(s) | InVivoPlus rat IgG2a isotype control, anti-trinitrophenol |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Recombinant PD-1-Ig fusion protein |
| Reported Applications |
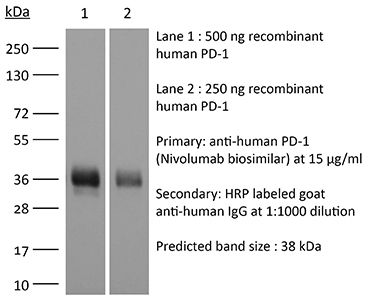
in vivo blocking of PD-1/PD-L signaling in vitro PD-1 neutralization Immunohistochemistry (frozen) Immunofluorescence Western blot Flow cytometry |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin* |
<1EU/mg (<0.001EU/μg) Determined by LAL gel clotting assay |
| Aggregation* |
<5% Determined by SEC |
| Purity |
>95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_2687796 |
| Molecular Weight | 150 kDa |
| Murine Pathogen Tests* |
Ectromelia/Mousepox Virus: Negative Hantavirus: Negative K Virus: Negative Lactate Dehydrogenase-Elevating Virus: Negative Lymphocytic Choriomeningitis virus: Negative Mouse Adenovirus: Negative Mouse Cytomegalovirus: Negative Mouse Hepatitis Virus: Negative Mouse Minute Virus: Negative Mouse Norovirus: Negative Mouse Parvovirus: Negative Mouse Rotavirus: Negative Mycoplasma Pulmonis: Negative Pneumonia Virus of Mice: Negative Polyoma Virus: Negative Reovirus Screen: Negative Sendai Virus: Negative Theiler’s Murine Encephalomyelitis: Negative |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
Additional Formats




Recommended Products
-

Recommended Isotype Control(s)
InVivoPlus rat IgG2a isotype control, anti-trinitrophenol
-

Recommended Dilution Buffer
InVivoPure pH 7.0 Dilution Buffer
in vivo blocking of PD-1/PD-L signaling
Wang, W., et al. (2018). "RIP1 Kinase Drives Macrophage-Mediated Adaptive Immune Tolerance in Pancreatic Cancer" Cancer Cell 34(5): 757-774 e757. PubMed
Pancreatic ductal adenocarcinoma (PDA) is characterized by immune tolerance and immunotherapeutic resistance. We discovered upregulation of receptor-interacting serine/threonine protein kinase 1 (RIP1) in tumor-associated macrophages (TAMs) in PDA. To study its role in oncogenic progression, we developed a selective small-molecule RIP1 inhibitor with high in vivo exposure. Targeting RIP1 reprogrammed TAMs toward an MHCII(hi)TNFalpha(+)IFNgamma(+) immunogenic phenotype in a STAT1-dependent manner. RIP1 inhibition in TAMs resulted in cytotoxic T cell activation and T helper cell differentiation toward a mixed Th1/Th17 phenotype, leading to tumor immunity in mice and in organotypic models of human PDA. Targeting RIP1 synergized with PD1-and inducible co-stimulator-based immunotherapies. Tumor-promoting effects of RIP1 were independent of its co-association with RIP3. Collectively, our work describes RIP1 as a checkpoint kinase governing tumor immunity.
in vivo blocking of PD-1/PD-L signaling
Gordon, S. R., et al. (2017). "PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity" Nature 545(7655): 495-499. PubMed
Programmed cell death protein 1 (PD-1) is an immune checkpoint receptor that is upregulated on activated T cells for the induction of immune tolerance. Tumour cells frequently overexpress the ligand for PD-1, programmed cell death ligand 1 (PD-L1), facilitating their escape from the immune system. Monoclonal antibodies that block the interaction between PD-1 and PD-L1, by binding to either the ligand or receptor, have shown notable clinical efficacy in patients with a variety of cancers, including melanoma, colorectal cancer, non-small-cell lung cancer and Hodgkin’s lymphoma. Although it is well established that PD-1-PD-L1 blockade activates T cells, little is known about the role that this pathway may have in tumour-associated macrophages (TAMs). Here we show that both mouse and human TAMs express PD-1. TAM PD-1 expression increases over time in mouse models of cancer and with increasing disease stage in primary human cancers. TAM PD-1 expression correlates negatively with phagocytic potency against tumour cells, and blockade of PD-1-PD-L1 in vivo increases macrophage phagocytosis, reduces tumour growth and lengthens the survival of mice in mouse models of cancer in a macrophage-dependent fashion. This suggests that PD-1-PD-L1 therapies may also function through a direct effect on macrophages, with substantial implications for the treatment of cancer with these agents.
in vivo blocking of PD-1/PD-L signaling, Flow Cytometry
Koyama, S., et al. (2016). "Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints" Nat Commun 7: 10501. PubMed
Despite compelling antitumour activity of antibodies targeting the programmed death 1 (PD-1): programmed death ligand 1 (PD-L1) immune checkpoint in lung cancer, resistance to these therapies has increasingly been observed. In this study, to elucidate mechanisms of adaptive resistance, we analyse the tumour immune microenvironment in the context of anti-PD-1 therapy in two fully immunocompetent mouse models of lung adenocarcinoma. In tumours progressing following response to anti-PD-1 therapy, we observe upregulation of alternative immune checkpoints, notably T-cell immunoglobulin mucin-3 (TIM-3), in PD-1 antibody bound T cells and demonstrate a survival advantage with addition of a TIM-3 blocking antibody following failure of PD-1 blockade. Two patients who developed adaptive resistance to anti-PD-1 treatment also show a similar TIM-3 upregulation in blocking antibody-bound T cells at treatment failure. These data suggest that upregulation of TIM-3 and other immune checkpoints may be targetable biomarkers associated with adaptive resistance to PD-1 blockade.
in vivo blocking of PD-1/PD-L signaling
Koyama, S., et al. (2016). "STK11/LKB1 Deficiency Promotes Neutrophil Recruitment and Proinflammatory Cytokine Production to Suppress T-cell Activity in the Lung Tumor Microenvironment" Cancer Res 76(5): 999-1008. PubMed
STK11/LKB1 is among the most commonly inactivated tumor suppressors in non-small cell lung cancer (NSCLC), especially in tumors harboring KRAS mutations. Many oncogenes promote immune escape, undermining the effectiveness of immunotherapies, but it is unclear whether the inactivation of tumor suppressor genes, such as STK11/LKB1, exerts similar effects. In this study, we investigated the consequences of STK11/LKB1 loss on the immune microenvironment in a mouse model of KRAS-driven NSCLC. Genetic ablation of STK11/LKB1 resulted in accumulation of neutrophils with T-cell-suppressive effects, along with a corresponding increase in the expression of T-cell exhaustion markers and tumor-promoting cytokines. The number of tumor-infiltrating lymphocytes was also reduced in LKB1-deficient mouse and human tumors. Furthermore, STK11/LKB1-inactivating mutations were associated with reduced expression of PD-1 ligand PD-L1 in mouse and patient tumors as well as in tumor-derived cell lines. Consistent with these results, PD-1-targeting antibodies were ineffective against Lkb1-deficient tumors. In contrast, treating Lkb1-deficient mice with an IL6-neutralizing antibody or a neutrophil-depleting antibody yielded therapeutic benefits associated with reduced neutrophil accumulation and proinflammatory cytokine expression. Our findings illustrate how tumor suppressor mutations can modulate the immune milieu of the tumor microenvironment, and they offer specific implications for addressing STK11/LKB1-mutated tumors with PD-1-targeting antibody therapies.
in vitro PD-1 neutralization
Park, S. J., et al. (2014). "Negative role of inducible PD-1 on survival of activated dendritic cells" J Leukoc Biol 95(4): 621-629. PubMed
PD-1 is a well-established negative regulator of T cell responses by inhibiting proliferation and cytokine production of T cells via interaction with its ligands, B7-H1 (PD-L1) and B7-DC (PD-L2), expressed on non-T cells. Recently, PD-1 was found to be expressed in innate cells, including activated DCs, and plays roles in suppressing production of inflammatory cytokines. In this study, we demonstrate that PD-1 KO DCs exhibited prolonged longevity compared with WT DCs in the dLNs after transfer of DCs into hind footpads. Interestingly, upon LPS stimulation, WT DCs increased the expression of PD-1 and started to undergo apoptosis. DCs, in spleen of LPS-injected PD-1 KO mice, were more resistant to LPS-mediated apoptosis in vivo than WT controls. Moreover, treatment of blocking anti-PD-1 mAb during DC maturation resulted in enhanced DC survival, suggesting that PD-1:PD-L interactions are involved in DC apoptosis. As a result, PD-1-deficient DCs augmented T cell responses in terms of antigen-specific IFN-gamma production and proliferation of CD4 and CD8 T cells to a greater degree than WT DCs. Moreover, PD-1 KO DCs exhibited increased MAPK1 and CD40-CD40L signaling, suggesting a possible mechanism for enhanced DC survival in the absence of PD-1 expression. Taken together, our findings further extend the function of PD-1, which plays an important role in apoptosis of activated DCs and provides important implications for PD-1-mediated immune regulation.
in vivo blocking of PD-1/PD-L signaling
Cooper, Z. A., et al. (2014). "Response to BRAF inhibition in melanoma is enhanced when combined with immune checkpoint blockade" Cancer Immunol Res 2(7): 643-654. PubMed
BRAF-targeted therapy results in objective responses in the majority of patients; however, the responses are short lived ( approximately 6 months). In contrast, treatment with immune checkpoint inhibitors results in a lower response rate, but the responses tend to be more durable. BRAF inhibition results in a more favorable tumor microenvironment in patients, with an increase in CD8(+) T-cell infiltrate and a decrease in immunosuppressive cytokines. There is also increased expression of the immunomodulatory molecule PDL1, which may contribute to the resistance. On the basis of these findings, we hypothesized that BRAF-targeted therapy may synergize with the PD1 pathway blockade to enhance antitumor immunity. To test this hypothesis, we developed a BRAF(V600E)/Pten(-/-) syngeneic tumor graft immunocompetent mouse model in which BRAF inhibition leads to a significant increase in the intratumoral CD8(+) T-cell density and cytokine production, similar to the effects of BRAF inhibition in patients. In this model, CD8(+) T cells were found to play a critical role in the therapeutic effect of BRAF inhibition. Administration of anti-PD1 or anti-PDL1 together with a BRAF inhibitor led to an enhanced response, significantly prolonging survival and slowing tumor growth, as well as significantly increasing the number and activity of tumor-infiltrating lymphocytes. These results demonstrate synergy between combined BRAF-targeted therapy and immune checkpoint blockade. Although clinical trials combining these two strategies are ongoing, important questions still remain unanswered. Further studies using this new melanoma mouse model may provide therapeutic insights, including optimal timing and sequence of therapy.
in vivo blocking of PD-1/PD-L signaling, in vitro PD-1 neutralization
Duraiswamy, J., et al. (2013). "Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors" Cancer Res 73(12): 3591-3603. PubMed
Tumor progression is facilitated by regulatory T cells (Treg) and restricted by effector T cells. In this study, we document parallel regulation of CD8(+) T cells and Foxp3(+) Tregs by programmed death-1 (PD-1, PDCD1). In addition, we identify an additional role of CTL antigen-4 (CTLA-4) inhibitory receptor in further promoting dysfunction of CD8(+) T effector cells in tumor models (CT26 colon carcinoma and ID8-VEGF ovarian carcinoma). Two thirds of CD8(+) tumor-infiltrating lymphocytes (TIL) expressed PD-1, whereas one third to half of CD8(+) TIL coexpressed PD-1 and CTLA-4. Double-positive (PD-1(+)CTLA-4(+)) CD8(+) TIL had characteristics of more severe dysfunction than single-positive (PD-1(+) or CTLA-4(+)) TIL, including an inability to proliferate and secrete effector cytokines. Blockade of both PD-1 and CTLA-4 resulted in reversal of CD8(+) TIL dysfunction and led to tumor rejection in two thirds of mice. Double blockade was associated with increased proliferation of antigen-specific effector CD8(+) and CD4(+) T cells, antigen-specific cytokine release, inhibition of suppressive functions of Tregs, and upregulation of key signaling molecules critical for T-cell function. When used in combination with GVAX vaccination (consisting of granulocyte macrophage colony-stimulating factor-expressing irradiated tumor cells), inhibitory pathway blockade induced rejection of CT26 tumors in 100% of mice and ID8-VEGF tumors in 75% of mice. Our study indicates that PD-1 signaling in tumors is required for both suppressing effector T cells and maintaining tumor Tregs, and that PD-1/PD-L1 pathway (CD274) blockade augments tumor inhibition by increasing effector T-cell activity, thereby attenuating Treg suppression.
Flow Cytometry
Good-Jacobson, K. L., et al. (2012). "CD80 expression on B cells regulates murine T follicular helper development, germinal center B cell survival, and plasma cell generation" J Immunol 188(9): 4217-4225. PubMed
Germinal center (GC) B cells and T follicular helper (T(FH)) cells interact in the production of high-affinity long-lived plasma cells (PCs) and memory B cells, although the mechanisms regulating the formation of these long-lived populations remain unclear. Because CD80 is one of the few markers shared by human and murine memory B cells, we investigated its role in the development of GCs, memory cells, and PCs. In CD80-deficient mice, fewer long-lived PCs were generated upon immunization compared with that in B6 controls. In concert, the absence of CD80 resulted in an increase in apoptotic GC B cells during the contraction phase of the GC. CD80(-/-) mice had fewer T(FH) cells compared with that of B6, and residual T(FH) cells failed to mature, with decreased ICOS and PD-1 expression and decreased synthesis of IL-21 mRNA. Mixed bone marrow chimeras demonstrated a B cell-intrinsic requirement for CD80 expression for normal T(FH) cell and PC development. Therefore, B cell expression of CD80 plays a critical role in regulating B-T interactions in both early and late GC responses. This, in turn, results in impaired ability to produce long-lived PCs. These data provide new insights into the development of GCs and Ab-forming cells and the functions of CD80 in humoral immunity.
Immunofluorescence, Western Blot
Chen, L., et al. (2009). "Role of the immune modulator programmed cell death-1 during development and apoptosis of mouse retinal ganglion cells" Invest Ophthalmol Vis Sci 50(10): 4941-4948. PubMed
PURPOSE: Mammalian programmed cell death (PD)-1 is a membrane-associated receptor regulating the balance between T-cell activation, tolerance, and immunopathology; however, its role in neurons has not yet been defined. The hypothesis that PD-1 signaling actively promotes retinal ganglion cell (RGC) death within the developing mouse retina was investigated. METHODS: Mature retinal cell types expressing PD-1 were identified by immunofluorescence staining of vertical retina sections; developmental expression was localized by immunostaining and quantified by Western blot analysis. PD-1 involvement in developmental RGC survival was assessed in vitro using retinal explants and in vivo using PD-1 knockout mice. PD-1 ligand gene expression was detected by RT-PCR. RESULTS: PD-1 is expressed in most adult RGCs and undergoes dynamic upregulation during the early postnatal window of retinal cell maturation and physiological programmed cell death (PCD). In vitro blockade of PD-1 signaling during this time selectively increases the survival of RGCs. Furthermore, PD-1-deficient mice show a selective increase in RGC number in the neonatal retina at the peak of developmental RGC death. Lastly, gene expression of the immune PD-1 ligand genes Pdcd1lg1 and Pdcd1lg2 was found throughout postnatal retina maturation. CONCLUSIONS: These findings collectively support a novel role for a PD-1-mediated signaling pathway in developmental PCD during postnatal RGC maturation.
Immunohistochemistry (frozen)
Menke, J., et al. (2007). "Programmed death 1 ligand (PD-L) 1 and PD-L2 limit autoimmune kidney disease: distinct roles" J Immunol 179(11): 7466-7477. PubMed
The programmed death 1/programmed death 1 ligand (PD-L) pathway is instrumental in peripheral tolerance. Blocking this pathway exacerbates experimental autoimmune diseases, but its role in autoimmune kidney disease has not been explored. Therefore, we tested the hypothesis that the programmed death 1 ligands (PD-L1 and PD-L2), provide a protective barrier during T cell- and macrophage (Mphi)-dependent autoimmune kidney disease. For this purpose, we compared nephrotoxic serum nephritis (NSN) in mice lacking PD-L1 (PD-L1(-/-)), PD-L2 (PD-L2(-/-)), or both (PD-L1/L2(-/-)) to wild-type (WT) C57BL/6 mice. Kidney pathology, loss of renal function, and intrarenal leukocyte infiltrates were increased in each PD-L(-/-) strain as compared with WT mice. Although the magnitude of renal pathology was similar in PD-L1(-/-) and PD-L2(-/-) mice, our findings suggest that kidney disease in each strain is regulated by distinct mechanisms. Specifically, we detected increased CD68(+) cells along with elevated circulating IgG and IgG deposits in glomeruli in PD-L2(-/-) mice, but not PD-L1(-/-) mice. In contrast, we detected a rise in activated CD8(+) T cells in PD-L1(-/-) mice, but not PD-L2(-/-) mice. Furthermore, since PD-L1 is expressed by parenchymal and hemopoietic cells in WT kidneys, we explored the differential impact of PD-L1 expression on these cell types by inducing NSN in bone marrow chimeric mice. Our results indicate that PD-L1 expression on hemopoietic cells, and not parenchymal cells, is primarily responsible for limiting leukocyte infiltration during NSN. Taken together, our findings indicate that PD-L1 and PD-L2 provide distinct negative regulatory checkpoints poised to suppress autoimmune renal disease.
in vivo blocking of PD-1/PD-L signaling
Barber, D. L., et al. (2006). "Restoring function in exhausted CD8 T cells during chronic viral infection" Nature 439(7077): 682-687. PubMed
Functional impairment of antigen-specific T cells is a defining characteristic of many chronic infections, but the underlying mechanisms of T-cell dysfunction are not well understood. To address this question, we analysed genes expressed in functionally impaired virus-specific CD8 T cells present in mice chronically infected with lymphocytic choriomeningitis virus (LCMV), and compared these with the gene profile of functional memory CD8 T cells. Here we report that PD-1 (programmed death 1; also known as Pdcd1) was selectively upregulated by the exhausted T cells, and that in vivo administration of antibodies that blocked the interaction of this inhibitory receptor with its ligand, PD-L1 (also known as B7-H1), enhanced T-cell responses. Notably, we found that even in persistently infected mice that were lacking CD4 T-cell help, blockade of the PD-1/PD-L1 inhibitory pathway had a beneficial effect on the ‘helpless’ CD8 T cells, restoring their ability to undergo proliferation, secrete cytokines, kill infected cells and decrease viral load. Blockade of the CTLA-4 (cytotoxic T-lymphocyte-associated protein 4) inhibitory pathway had no effect on either T-cell function or viral control. These studies identify a specific mechanism of T-cell exhaustion and define a potentially effective immunological strategy for the treatment of chronic viral infections.
Immunohistochemistry (frozen)
Liang, S. C., et al. (2003). "Regulation of PD-1, PD-L1, and PD-L2 expression during normal and autoimmune responses" Eur J Immunol 33(10): 2706-2716. PubMed
Newer members of the B7-CD28 superfamily include the receptor PD-1 and its two ligands, PD-L1 and PD-L2. Here, we characterize the expression of PD-1, PD-L1, and PD-L2 in tissues of naive miceand in target organs from two models of autoimmunity, the pancreas from non-obese diabetic (NOD) mice and brain from mice with experimental autoimmune encephalomyelitis (EAE). In naive mice, proteiexpression of PD-1, PD-L1, and PD-L2 was detected in the thymus, while PD-1 and PD-L1 were detected in the spleen. PD-L1, but not PD-L2, was also detected at low levels on cardiac endothelium, pancreatic islets, and syncyciotrophoblasts in the placenta. In pre-diabetic NOD mice, PD-1 and PD-L1 were expressed on infiltrating cells in the pancreatic islets. Furthermore, PD-L1 was markedly up-regulated on islet cells. In brains from mice with EAE, PD-1, PD-L1, and PD-L2 were expressed on infiltrating inflammatory cells, and PD-L1 was up-regulated on endothelium within EAE brain. The distinct expression patterns of PD-L1 and PD-L2 led us to compare their transcriptional regulation in STAT4(-/-), STAT6(-/-), or NF-kappaB p50(-/-)p65(+/-) dendritic cells (DC).PD-L2, but not PD-L1, expression was dramatically reduced in p50(-/-)p65(+/-) DC. Thus, PD-L1 and PD-L2 exhibit distinct expression patterns and are differentially regulated on the transcriptional level.