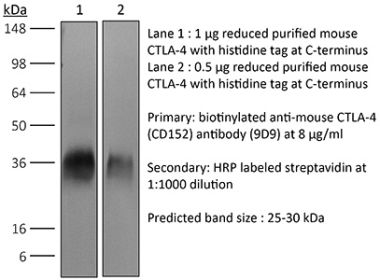
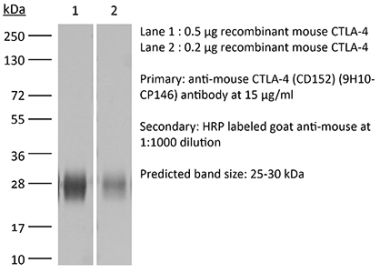
InVivoPlus anti-mouse CTLA-4 (CD152)

Product Details
The UC10-4F10-11 monoclonal antibody reacts with mouse CTLA-4 (cytotoxic T lymphocyte antigen-4) also known as CD152. CTLA-4 is a 33 kDa cell surface receptor encoded by the Ctla4 gene that belongs to the CD28 family of the Ig superfamily. CTLA-4 is expressed on activated T and B lymphocytes. CTLA-4 is structurally similar to the T-cell co-stimulatory protein, CD28, and both molecules bind to the B7 family members B7-1 (CD80) and B7-2 (CD86). Upon ligand binding, CTLA-4 negatively regulates cell-mediated immune responses. CTLA-4 plays roles in induction and/or maintenance of immunological tolerance, thymocyte development, and regulation of protective immunity. The critical role of CTLA-4 in immune down-regulation has been demonstrated in CTLA-4 deficient mice, which succumb at 3-5 weeks of age due to the development of a lymphoproliferative disease. CTLA-4 is among a group of inhibitory receptors being explored as cancer treatment targets through immune checkpoint blockade. The UC10-4F10-11 antibody has been shown to promote T cell co-stimulation by blocking CTLA-4 binding to the B7 co-receptors, allowing for CD28 binding.Specifications
| Isotype | Armenian hamster IgG |
|---|---|
| Recommended Isotype Control(s) | InVivoPlus polyclonal Armenian hamster IgG |
| Recommended Dilution Buffer | InVivoPure pH 6.5 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Mouse CTLA-4 IgG2a fusion protein |
| Reported Applications |
in vivo CTLA-4 neutralization in vitro CTLA-4 neutralization Flow cytometry Western blot |
| Formulation |
PBS, pH 6.5 Contains no stabilizers or preservatives |
| Endotoxin* |
<1EU/mg (<0.001EU/μg) Determined by LAL gel clotting assay |
| Aggregation* |
<5% Determined by SEC |
| Purity |
>95% Determined by SDS-PAGE |
| Sterility | 0.2 μM filtered |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_1107598 |
| Molecular Weight | 150 kDa |
| Murine Pathogen Tests* |
Ectromelia/Mousepox Virus: Negative Hantavirus: Negative K Virus: Negative Lactate Dehydrogenase-Elevating Virus: Negative Lymphocytic Choriomeningitis virus: Negative Mouse Adenovirus: Negative Mouse Cytomegalovirus: Negative Mouse Hepatitis Virus: Negative Mouse Minute Virus: Negative Mouse Norovirus: Negative Mouse Parvovirus: Negative Mouse Rotavirus: Negative Mycoplasma Pulmonis: Negative Pneumonia Virus of Mice: Negative Polyoma Virus: Negative Reovirus Screen: Negative Sendai Virus: Negative Theiler’s Murine Encephalomyelitis: Negative |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
Additional Formats

Recommended Products
-

Recommended Isotype Control(s)
InVivoPlus polyclonal Armenian hamster IgG
-

Recommended Dilution Buffer
InVivoPure pH 6.5 Dilution Buffer
in vivo CTLA-4 neutralization
Triplett, T. A., et al. (2018). "Reversal of indoleamine 2,3-dioxygenase-mediated cancer immune suppression by systemic kynurenine depletion with a therapeutic enzyme" Nat Biotechnol 36(8): 758-764. PubMed
Increased tryptophan (Trp) catabolism in the tumor microenvironment (TME) can mediate immune suppression by upregulation of interferon (IFN)-gamma-inducible indoleamine 2,3-dioxygenase (IDO1) and/or ectopic expression of the predominantly liver-restricted enzyme tryptophan 2,3-dioxygenase (TDO). Whether these effects are due to Trp depletion in the TME or mediated by the accumulation of the IDO1 and/or TDO (hereafter referred to as IDO1/TDO) product kynurenine (Kyn) remains controversial. Here we show that administration of a pharmacologically optimized enzyme (PEGylated kynureninase; hereafter referred to as PEG-KYNase) that degrades Kyn into immunologically inert, nontoxic and readily cleared metabolites inhibits tumor growth. Enzyme treatment was associated with a marked increase in the tumor infiltration and proliferation of polyfunctional CD8(+) lymphocytes. We show that PEG-KYNase administration had substantial therapeutic effects when combined with approved checkpoint inhibitors or with a cancer vaccine for the treatment of large B16-F10 melanoma, 4T1 breast carcinoma or CT26 colon carcinoma tumors. PEG-KYNase mediated prolonged depletion of Kyn in the TME and reversed the modulatory effects of IDO1/TDO upregulation in the TME.
in vivo CTLA-4 neutralization, Flow Cytometry
Pletinckx, K., et al. (2015). "Immature dendritic cells convert anergic nonregulatory T cells into Foxp3- IL-10+ regulatory T cells by engaging CD28 and CTLA-4" Eur J Immunol 45(2): 480-491. PubMed
Anergic T cells can survive for long time periods passively in a hyporesponsive state without obvious active functions. Thus, the immunological reason for their maintenance is unclear. Here, we induced peptide-specific anergy in T cells from mice by coculturing these cells with immature murine dendritic cells (DCs). We found that these anergic, nonsuppressive IL-10(-) Foxp3(-) CTLA-4(+) CD25(low) Egr2(+) T cells could be converted into suppressive IL-10(+) Foxp3(-) CTLA-4(+) CD25(high) Egr2(+) cells resembling type-1 Treg cells (Tr1) when stimulated a second time by immature DCs in vitro. Addition of TGF-beta during anergy induction favored Foxp3(+) Treg-cell induction, while TGF-beta had little effect when added to the second stimulation. Expression of both CD28 and CTLA-4 molecules on anergic T cells was required to allow their conversion into Tr1-like cells. Suppressor activity was enabled via CD28-mediated CD25 upregulation, acting as an IL-2 sink, together with a CTLA-4-mediated inhibition of NFATc1/alpha activation to shut down IL-2-mediated proliferation. Together, these data provide evidence and mechanistical insights into how persistent anergic T cells may serve as a resting memory pool for Tr1-like cells.
in vivo CTLA-4 neutralization
Welten, S. P., et al. (2015). "The viral context instructs the redundancy of costimulatory pathways in driving CD8(+) T cell expansion" Elife 4. doi : 10.7554/eLife.07486. PubMed
Signals delivered by costimulatory molecules are implicated in driving T cell expansion. The requirements for these signals, however, vary from dispensable to essential in different infections. We examined the underlying mechanisms of this differential T cell costimulation dependence and found that the viral context determined the dependence on CD28/B7-mediated costimulation for expansion of naive and memory CD8(+) T cells, indicating that the requirement for costimulatory signals is not imprinted. Notably, related to the high-level costimulatory molecule expression induced by lymphocytic choriomeningitis virus (LCMV), CD28/B7-mediated costimulation was dispensable for accumulation of LCMV-specific CD8(+) T cells because of redundancy with the costimulatory pathways induced by TNF receptor family members (i.e., CD27, OX40, and 4-1BB). Type I IFN signaling in viral-specific CD8(+) T cells is slightly redundant with costimulatory signals. These results highlight that pathogen-specific conditions differentially and uniquely dictate the utilization of costimulatory pathways allowing shaping of effector and memory antigen-specific CD8(+) T cell responses.
in vivo CTLA-4 neutralization
Makkouk, A., et al. (2015). "Three steps to breaking immune tolerance to lymphoma: a microparticle approach" Cancer Immunol Res 3(4): 389-398. PubMed
In situ immunization aims at generating antitumor immune responses through manipulating the tumor microenvironment. On the basis of recent advances in the understanding of antitumor immunity, we designed a three-step approach to in situ immunization to lymphoma: (i) inducing immunogenic tumor cell death with the chemotherapeutic drug doxorubicin. Doxorubicin enhances the expression of “eat-me” signals by dying tumor cells, facilitating their phagocytosis by dendritic cells (DC). Because of the vesicant activity of doxorubicin, microparticles made of biodegradable polymer poly(lactide-co-glycolide) or PLGA can safely deliver doxorubicin intratumorally and are effective vaccine adjuvants, (ii) enhancing T-cell activation using anti-OX40 and (iii) sustaining T-cell responses by checkpoint blockade using anti-CTLA-4. In vitro, doxorubicin microparticles were less cytotoxic to DCs than to B lymphoma cells, did not require internalization by tumor cells, and significantly enhanced phagocytosis of tumor cells by DCs as compared with soluble doxorubicin. In mice, this three-step therapy induced CD4- and CD8-dependent systemic immune responses that enhanced T-cell infiltration into distant tumors, leading to their eradication and significantly improving survival. Our findings demonstrate that systemic antitumor immune responses can be generated locally by three-step therapy and merit further investigation as an immunotherapy for patients with lymphoma.
in vivo CTLA-4 neutralization
Mittal, D., et al. (2014). "Antimetastatic effects of blocking PD-1 and the adenosine A2A receptor" Cancer Res 74(14): 3652-3658. PubMed
Adenosine targeting is an attractive new approach to cancer treatment, but no clinical study has yet examined adenosine inhibition in oncology despite the safe clinical profile of adenosine A2A receptor inhibitors (A2ARi) in Parkinson disease. Metastasis is the main cause of cancer-related deaths worldwide, and therefore we have studied experimental and spontaneous mouse models of melanoma and breast cancer metastasis to demonstrate the efficacy and mechanism of a combination of A2ARi in combination with anti-PD-1 monoclonal antibody (mAb). This combination significantly reduces metastatic burden and prolongs the life of mice compared with either monotherapy alone. Importantly, the combination was only effective when the tumor expressed high levels of CD73, suggesting a tumor biomarker that at a minimum could be used to stratify patients that might receive this combination. The mechanism of the combination therapy was critically dependent on NK cells and IFNgamma, and to a lesser extent, CD8(+) T cells and the effector molecule, perforin. Overall, these results provide a strong rationale to use A2ARi with anti-PD-1 mAb for the treatment of minimal residual and metastatic disease.
in vivo CTLA-4 neutralization
Zhu, Y., et al. (2014). "CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models" Cancer Res 74(18): 5057-5069. PubMed
Cancer immunotherapy generally offers limited clinical benefit without coordinated strategies to mitigate the immunosuppressive nature of the tumor microenvironment. Critical drivers of immune escape in the tumor microenvironment include tumor-associated macrophages and myeloid-derived suppressor cells, which not only mediate immune suppression, but also promote metastatic dissemination and impart resistance to cytotoxic therapies. Thus, strategies to ablate the effects of these myeloid cell populations may offer great therapeutic potential. In this report, we demonstrate in a mouse model of pancreatic ductal adenocarcinoma (PDAC) that inhibiting signaling by the myeloid growth factor receptor CSF1R can functionally reprogram macrophage responses that enhance antigen presentation and productive antitumor T-cell responses. Investigations of this response revealed that CSF1R blockade also upregulated T-cell checkpoint molecules, including PDL1 and CTLA4, thereby restraining beneficial therapeutic effects. We found that PD1 and CTLA4 antagonists showed limited efficacy as single agents to restrain PDAC growth, but that combining these agents with CSF1R blockade potently elicited tumor regressions, even in larger established tumors. Taken together, our findings provide a rationale to reprogram immunosuppressive myeloid cell populations in the tumor microenvironment under conditions that can significantly empower the therapeutic effects of checkpoint-based immunotherapeutics.
in vivo CTLA-4 neutralization
Rabenstein, H., et al. (2014). "Differential kinetics of antigen dependency of CD4+ and CD8+ T cells" J Immunol 192(8): 3507-3517. PubMed
Ag recognition via the TCR is necessary for the expansion of specific T cells that then contribute to adaptive immunity as effector and memory cells. Because CD4+ and CD8+ T cells differ in terms of their priming APCs and MHC ligands we compared their requirements of Ag persistence during their expansion phase side by side. Proliferation and effector differentiation of TCR transgenic and polyclonal mouse T cells were thus analyzed after transient and continuous TCR signals. Following equally strong stimulation, CD4+ T cell proliferation depended on prolonged Ag presence, whereas CD8+ T cells were able to divide and differentiate into effector cells despite discontinued Ag presentation. CD4+ T cell proliferation was neither affected by Th lineage or memory differentiation nor blocked by coinhibitory signals or missing inflammatory stimuli. Continued CD8+ T cell proliferation was truly independent of self-peptide/MHC-derived signals. The subset divergence was also illustrated by surprisingly broad transcriptional differences supporting a stronger propensity of CD8+ T cells to programmed expansion. These T cell data indicate an intrinsic difference between CD4+ and CD8+ T cells regarding the processing of TCR signals for proliferation. We also found that the presentation of a MHC class II-restricted peptide is more efficiently prolonged by dendritic cell activation in vivo than a class I bound one. In summary, our data demonstrate that CD4+ T cells require continuous stimulation for clonal expansion, whereas CD8+ T cells can divide following a much shorter TCR signal.
in vivo CTLA-4 neutralization, Flow Cytometry
Honda, T., et al. (2014). "Tuning of antigen sensitivity by T cell receptor-dependent negative feedback controls T cell effector function in inflamed tissues" Immunity 40(2): 235-247. PubMed
Activated T cells must mediate effector responses sufficiently to clear pathogens while avoiding excessive tissue damage. Here we have combined dynamic intravital microscopy with ex vivo assessments of T cell cytokine responses to generate a detailed spatiotemporal picture of CD4(+) T cell effector regulation in the skin. In response to antigen, effector T cells arrested transiently on antigen-presenting cells, briefly producing cytokine and then resuming migration. Antigen recognition led to upregulation of the programmed death-1 (PD-1) glycoprotein by T cells and blocking its canonical ligand, programmed death-ligand 1 (PD-L1), lengthened the duration of migration arrest and cytokine production, showing that PD-1 interaction with PD-L1 is a major negative feedback regulator of antigen responsiveness. We speculate that the immune system employs T cell recruitment, transient activation, and rapid desensitization to allow the T cell response to rapidly adjust to changes in antigen presentation and minimize collateral injury to the host.
in vivo CTLA-4 neutralization
Allard, B., et al. (2013). "Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs" Clin Cancer Res 19(20): 5626-5635. PubMed
PURPOSE: Monoclonal antibodies (mAb) that block programmed death (PD)-1 or cytotoxic T lymphocyte antigen (CTLA-4) receptors have been associated with durable clinical responses against a variety of cancer types and hold great potential as novel cancer therapeutics. Recent evidence suggest that targeted blockade of multiple immunosuppressive pathways can induce synergistic antitumor responses. EXPERIMENTAL DESIGN: In this study, we investigated whether targeted blockade of CD73, an ectonucleotidase that catabolizes the hydrolysis of extracellular adenosine monophosphate (AMP) to adenosine, can enhance the antitumor activity of anti-CTLA-4 and anti-PD-1 mAbs against transplanted and chemically induced mouse tumors. RESULTS: Anti-CD73 mAb significantly enhanced the activity of both anti-CTLA-4 and anti-PD-1 mAbs against MC38-OVA (colon) and RM-1 (prostate) subcutaneous tumors, and established metastatic 4T1.2 breast cancer. Anti-CD73 mAb also significantly enhanced the activity of anti-PD-1 mAb against 3-methylcholanthrene (MCA)-induced fibrosarcomas. Gene-targeted mice revealed that single-agent therapies and combinatorial treatments were dependent on host IFN-gamma and CD8(+) T cells, but independent of perforin. Interestingly, anti-CD73 mAb preferentially synergized with anti-PD-1 mAb. We investigated the effect of extracellular adenosine on tumor-infiltrating T cells and showed that activation of A2A adenosine receptor enhances PD-1 expression, but not CTLA-4 expression, on tumor-specific CD8+ T cells and CD4+ Foxp3+ T regulatory cells. CONCLUSIONS: Taken together, our study revealed that targeted blockade of CD73 can enhance the therapeutic activity of anti-PD-1 and anti-CTLA-4 mAbs and may thus potentiate therapeutic strategies targeting immune checkpoint inhibitors in general.
in vivo CTLA-4 neutralization
Hafalla, J. C., et al. (2012). "The CTLA-4 and PD-1/PD-L1 inhibitory pathways independently regulate host resistance to Plasmodium-induced acute immune pathology" PLoS Pathog 8(2): e1002504. PubMed
The balance between pro-inflammatory and regulatory immune responses in determining optimal T cell activation is vital for the successful resolution of microbial infections. This balance is maintained in part by the negative regulators of T cell activation, CTLA-4 and PD-1/PD-L, which dampen effector responses during chronic infections. However, their role in acute infections, such as malaria, remains less clear. In this study, we determined the contribution of CTLA-4 and PD-1/PD-L to the regulation of T cell responses during Plasmodium berghei ANKA (PbA)-induced experimental cerebral malaria (ECM) in susceptible (C57BL/6) and resistant (BALB/c) mice. We found that the expression of CTLA-4 and PD-1 on T cells correlates with the extent of pro-inflammatory responses induced during PbA infection, being higher in C57BL/6 than in BALB/c mice. Thus, ECM develops despite high levels of expression of these inhibitory receptors. However, antibody-mediated blockade of either the CTLA-4 or PD-1/PD-L1, but not the PD-1/PD-L2, pathways during PbA-infection in ECM-resistant BALB/c mice resulted in higher levels of T cell activation, enhanced IFN-gamma production, increased intravascular arrest of both parasitised erythrocytes and CD8(+) T cells to the brain, and augmented incidence of ECM. Thus, in ECM-resistant BALB/c mice, CTLA-4 and PD-1/PD-L1 represent essential, independent and non-redundant pathways for maintaining T cell homeostasis during a virulent malaria infection. Moreover, neutralisation of IFN-gamma or depletion of CD8(+) T cells during PbA infection was shown to reverse the pathologic effects of regulatory pathway blockade, highlighting that the aetiology of ECM in the BALB/c mice is similar to that in C57BL/6 mice. In summary, our results underscore the differential and complex regulation that governs immune responses to malaria parasites.
in vivo CTLA-4 neutralization
Haque, A., et al. (2010). "CD4+ natural regulatory T cells prevent experimental cerebral malaria via CTLA-4 when expanded in vivo" PLoS Pathog 6(12): e1001221. PubMed
Studies in malaria patients indicate that higher frequencies of peripheral blood CD4(+) Foxp3(+) CD25(+) regulatory T (Treg) cells correlate with increased blood parasitemia. This observation implies that Treg cells impair pathogen clearance and thus may be detrimental to the host during infection. In C57BL/6 mice infected with Plasmodium berghei ANKA, depletion of Foxp3(+) cells did not improve parasite control or disease outcome. In contrast, elevating frequencies of natural Treg cells in vivo using IL-2/anti-IL-2 complexes resulted in complete protection against severe disease. This protection was entirely dependent upon Foxp3(+) cells and resulted in lower parasite biomass, impaired antigen-specific CD4(+) T and CD8(+) T cell responses that would normally promote parasite tissue sequestration in this model, and reduced recruitment of conventional T cells to the brain. Furthermore, Foxp3(+) cell-mediated protection was dependent upon CTLA-4 but not IL-10. These data show that T cell-mediated parasite tissue sequestration can be reduced by regulatory T cells in a mouse model of malaria, thereby limiting malaria-induced immune pathology.
in vivo CTLA-4 neutralization
Rowe, J. H., et al. (2009). "Cytotoxic T-lymphocyte antigen 4 blockade augments the T-cell response primed by attenuated Listeria monocytogenes resulting in more rapid clearance of virulent bacterial challenge" Immunology 128(1 Suppl): e471-478. PubMed
Cytotoxic T-lymphocyte antigen 4 (CTLA-4) uniformly suppresses antigen-specific T cells during chronic infection with bacterial, parasitic or viral pathogens. However, the importance of CTLA-4 in controlling the T-cell response during acute infection or after priming with live attenuated vaccine vectors has not been well characterized. Since strategies aimed at blocking CTLA-4 are being actively developed to therapeutically augment T-cell-mediated immunity, the effects of CTLA-4 blockade on T-cell activation during these conditions need to be more clearly defined. We have examined the role of CTLA-4 in a prime-challenge model of acute bacterial infection using both attenuated and virulent strains of the intracellular bacterium Listeria monocytogenes. Although Foxp3(+) CD4(+) T cells are the predominant CTLA-4-expressing cell type in naive mice, antigen-specific Foxp3(-) CD4(+) cells upregulate CTLA-4 expression after primary L. monocytogenes infection. Blockade of CTLA-4 results in increased numbers of L. monocytogenes-specific CD4 and CD8 T cells after primary infection with attenuated L. monocytogenes, and confers more rapid bacterial clearance after secondary challenge with virulent L. monocytogenes. Accordingly, CTLA-4 plays an important suppressive role in T-cell priming and protective immunity in a prime-challenge model of acute bacterial infection.
in vivo CTLA-4 neutralization, in vitro CTLA-4 neutralization
Noval Rivas, M., et al. (2009). "Reviving function in CD4+ T cells adapted to persistent systemic antigen" J Immunol 183(7): 4284-4291. PubMed
In bone marrow-transplanted patients, chronic graft-versus-host disease is a complication that results from the persistent stimulation of recipient minor histocompatibility Ag (mHA)-specific T cells contained within the graft. In this study, we developed a mouse model where persistent stimulation of donor T cells by recipient’s mHA led to multiorgan T cell infiltration. Exposure to systemic mHA, however, deeply modified T cell function and chronically stimulated T cells developed a long-lasting state of unresponsiveness, or immune adaptation, characterized by their inability to mediate organ immune damages in vivo. However, analysis of the gene expression profile of adapted CD4+ T cells revealed the specific coexpression of genes known to promote differentiation and function of Th1 effector cells as well as genes coding for proteins that control T cell activity, such as cell surface-negative costimulatory molecules and regulatory cytokines. Strikingly, blockade of negative costimulation abolished T cell adaptation and stimulated strong IFN-gamma production and severe multiorgan wasting disease. Negative costimulation was also shown to control lethal LPS-induced toxic shock in mice with adapted T cells, as well as the capacity of adapted T cells to reject skin graft. Our results demonstrate that negative costimulation is the molecular mechanism used by CD4+ T cells to adapt their activity in response to persistent antigenic stimulation. The effector function of CD4+ T cells that have adapted to chronic Ag presentation can be activated by stimuli strong enough to overcome regulatory signals delivered to the T cells by negative costimulation.