InVivoPlus anti-mouse TNFα

Product Details
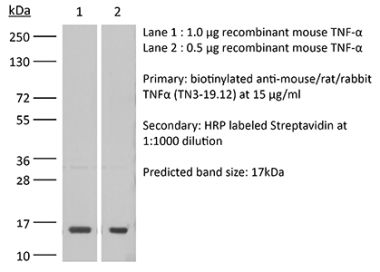
The XT3.11 monoclonal antibody reacts with mouse TNFα (tumor necrosis factor-alpha) a multifunctional proinflammatory cytokine. TNFα exists as a soluble 17 kDa monomer, which forms homotrimers in circulation or as a 26 kDa membrane-bound form. TNFα belongs to the TNF superfamily of cytokines and signals through its two receptors, TNFR1 and TNFR2 which can be activated by both the soluble trimeric and membrane-bound and forms of TNFα. TNFα is primarily produced by macrophages in response to foreign antigens such as bacteria (lipopolysaccharides), viruses, and parasites as well as mitogens and other cytokines but can also be expressed by monocytes, neutrophils, NK cells, CD4 T cells and some specialized dendritic cells. TNFα is known to play key roles in a wide spectrum of biological processes including immunoregulation, cell proliferation, differentiation, apoptosis, antitumor activity, inflammation, anorexia, cachexia, septic shock, hematopoiesis, and viral replication. TNFα dysregulation has been implicated in a variety of diseases, including autoimmune diseases, insulin resistance, and cancer. Mouse and human TNFα share 79% amino acid sequence identity however, mouse TNFα is glycosylated while human TNFα is not. TNFα knockout animals display defects in response to bacterial infection, characterized by defects in forming organized follicular dendritic cell networks and germinal centers with a lack of primary B cell follicles.Specifications
| Isotype | Rat IgG1 |
|---|---|
| Recommended Isotype Control(s) | InVivoPlus rat IgG1 isotype control, anti-horseradish peroxidase |
| Recommended Dilution Buffer | InVivoPure pH 8.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Recombinant mouse TNFα |
| Reported Applications |
in vivo TNFα neutralization in vitro TNFα neutralization Western blot |
| Formulation |
PBS, pH 8.0 Contains no stabilizers or preservatives |
| Endotoxin* |
<1EU/mg (<0.001EU/μg) Determined by LAL gel clotting assay |
| Aggregation* |
<5% Determined by SEC |
| Purity |
>95% Determined by SDS-PAGE |
| Sterility | 0.2 μM filtered |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_1107764 |
| Molecular Weight | 150 kDa |
| Murine Pathogen Tests* |
Ectromelia/Mousepox Virus: Negative Hantavirus: Negative K Virus: Negative Lactate Dehydrogenase-Elevating Virus: Negative Lymphocytic Choriomeningitis virus: Negative Mouse Adenovirus: Negative Mouse Cytomegalovirus: Negative Mouse Hepatitis Virus: Negative Mouse Minute Virus: Negative Mouse Norovirus: Negative Mouse Parvovirus: Negative Mouse Rotavirus: Negative Mycoplasma Pulmonis: Negative Pneumonia Virus of Mice: Negative Polyoma Virus: Negative Reovirus Screen: Negative Sendai Virus: Negative Theiler’s Murine Encephalomyelitis: Negative |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
Additional Formats

Recommended Products
-

Recommended Isotype Control(s)
InVivoPlus rat IgG1 isotype control, anti-horseradish peroxidase
-

Recommended Dilution Buffer
InVivoPure pH 8.0 Dilution Buffer
in vivo TNFα neutralization
Shaabani, N., et al. (2018). "The probacterial effect of type I interferon signaling requires its own negative regulator USP18" Sci Immunol 3(27). PubMed
Type I interferon (IFN-I) signaling paradoxically impairs host immune responses during many primary and secondary bacterial infections. Lack of IFN-I receptor reduces bacterial replication and/or bacterial persistence during infection with several bacteria. However, the mechanisms that mediate the adverse IFN-I effect are incompletely understood. Here, we show that Usp18, an interferon-stimulated gene that negatively regulates IFN-I signaling, is primarily responsible for the deleterious effect of IFN-I signaling during infection of mice with Listeria monocytogenes or Staphylococcus aureus Mechanistically, USP18 promoted bacterial replication by inhibiting antibacterial tumor necrosis factor-alpha (TNF-alpha) signaling. Deleting IFNAR1 or USP18 in CD11c-Cre(+) cells similarly reduced bacterial titers in multiple organs and enhanced survival. Our results demonstrate that inhibiting USP18 function can promote control of primary and secondary bacterial infection by enhancing the antibacterial effect of TNF-alpha, which correlates with induction of reactive oxygen species (ROS). These findings suggest that USP18 could be targeted therapeutically in patients to ameliorate disease caused by serious bacterial infections.
in vivo TNFα neutralization
Grinberg-Bleyer, Y., et al. (2015). "Cutting edge: NF-kappaB p65 and c-Rel control epidermal development and immune homeostasis in the skin" J Immunol 194(6): 2472-2476. PubMed
Psoriasis is an inflammatory skin disease in which activated immune cells and the proinflammatory cytokine TNF are well-known mediators of pathogenesis. The transcription factor NF-kappaB is a key regulator of TNF production and TNF-induced proinflammatory gene expression, and both the psoriatic transcriptome and genetic susceptibility further implicate NF-kappaB in psoriasis etiopathology. However, the role of NF-kappaB in psoriasis remains controversial. We analyzed the function of canonical NF-kappaB in the epidermis using CRE-mediated deletion of p65 and c-Rel in keratinocytes. In contrast to animals lacking p65 or c-Rel alone, mice lacking both subunits developed severe dermatitis after birth. Consistent with its partial histological similarity to human psoriasis, this condition could be prevented by anti-TNF treatment. Moreover, regulatory T cells in lesional skin played an important role in disease remission. Our results demonstrate that canonical NF-kappaB in keratinocytes is essential for the maintenance of skin immune homeostasis and is protective against spontaneous dermatitis.
in vivo TNFα neutralization
Christensen, A. D., et al. (2015). "Depletion of regulatory T cells in a hapten-induced inflammation model results in prolonged and increased inflammation driven by T cells" Clin Exp Immunol 179(3): 485-499. PubMed
Regulatory T cells (Tregs ) are known to play an immunosuppressive role in the response of contact hypersensitivity (CHS), but neither the dynamics of Tregs during the CHS response nor the exaggerated inflammatory response after depletion of Tregs has been characterized in detail. In this study we show that the number of Tregs in the challenged tissue peak at the same time as the ear-swelling reaches its maximum on day 1 after challenge, whereas the number of Tregs in the draining lymph nodes peaks at day 2. As expected, depletion of Tregs by injection of a monoclonal antibody to CD25 prior to sensitization led to a prolonged and sustained inflammatory response which was dependent upon CD8 T cells, and co-stimulatory blockade with cytotoxic T lymphocyte antigen-4-immunoglobulin (CTLA-4-Ig) suppressed the exaggerated inflammation. In contrast, blockade of the interleukin (IL)-10-receptor (IL-10R) did not further increase the exaggerated inflammatory response in the Treg -depleted mice. In the absence of Tregs , the response changed from a mainly acute reaction with heavy infiltration of neutrophils to a sustained response with more chronic characteristics (fewer neutrophils and dominated by macrophages). Furthermore, depletion of Tregs enhanced the release of cytokines and chemokines locally in the inflamed ear and augmented serum levels of the systemic inflammatory mediators serum amyloid (SAP) and haptoglobin early in the response.
in vivo TNFα neutralization
Baeyens, A., et al. (2015). "Effector T cells boost regulatory T cell expansion by IL-2, TNF, OX40, and plasmacytoid dendritic cells depending on the immune context" J Immunol 194(3): 999-1010. PubMed
CD4(+)CD25(+)Foxp3(+) regulatory T (Treg) cells play a major role in peripheral tolerance. Multiple environmental factors and cell types affect their biology. Among them, activated effector CD4(+) T cells can boost Treg cell expansion through TNF or IL-2. In this study, we further characterized this effector T (Teff) cell-dependent Treg cell boost in vivo in mice. This phenomenon was observed when both Treg and Teff cells were activated by their cognate Ag, with the latter being the same or different. Also, when Treg cells highly proliferated on their own, there was no additional Treg cell boost by Teff cells. In a condition of low inflammation, the Teff cell-mediated Treg cell boost involved TNF, OX40L, and plasmacytoid dendritic cells, whereas in a condition of high inflammation, it involved TNF and IL-2. Thus, this feedback mechanism in which Treg cells are highly activated by their Teff cell counterparts depends on the immune context for its effectiveness and mechanism. This Teff cell-dependent Treg cell boost may be crucial to limit inflammatory and autoimmune responses.
in vivo TNFα neutralization
Yoo, J. K. and T. J. Braciale. (2014). "IL-21 promotes late activator APC-mediated T follicular helper cell differentiation in experimental pulmonary virus infection" PLoS One 9(9): e105872. PubMed
IL-21 is a type-I cytokine that has pleiotropic immuno-modulatory effects. Primarily produced by activated T cells including NKT and TFH cells, IL-21 plays a pivotal role in promoting TFH differentiation through poorly understood cellular and molecular mechanisms. Here, employing a mouse model of influenza A virus (IAV) infection, we demonstrate that IL-21, initially produced by NKT cells, promotes TFH differentiation by promoting the migration of late activator antigen presenting cell (LAPC), a recently identified TFH inducer, from the infected lungs into the draining lymph nodes (dLN). LAPC migration from IAV-infected lung into the dLN is CXCR3-CXCL9 dependent. IL-21-induced TNF-alpha production by conventional T cells is critical to stimulate CXCL9 expression by DCs in the dLN, which supports LAPC migration into the dLN and ultimately facilitates TFH differentiation. Our results reveal a previously unappreciated mechanism for IL-21 modulation of TFH responses during respiratory virus infection.
in vivo TNFα neutralization
Deng, L., et al. (2014). "Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice" J Clin Invest 124(2): 687-695. PubMed
High-dose ionizing irradiation (IR) results in direct tumor cell death and augments tumor-specific immunity, which enhances tumor control both locally and distantly. Unfortunately, local relapses often occur following IR treatment, indicating that IR-induced responses are inadequate to maintain antitumor immunity. Therapeutic blockade of the T cell negative regulator programmed death-ligand 1 (PD-L1, also called B7-H1) can enhance T cell effector function when PD-L1 is expressed in chronically inflamed tissues and tumors. Here, we demonstrate that PD-L1 was upregulated in the tumor microenvironment after IR. Administration of anti-PD-L1 enhanced the efficacy of IR through a cytotoxic T cell-dependent mechanism. Concomitant with IR-mediated tumor regression, we observed that IR and anti-PD-L1 synergistically reduced the local accumulation of tumor-infiltrating myeloid-derived suppressor cells (MDSCs), which suppress T cells and alter the tumor immune microenvironment. Furthermore, activation of cytotoxic T cells with combination therapy mediated the reduction of MDSCs in tumors through the cytotoxic actions of TNF. Our data provide evidence for a close interaction between IR, T cells, and the PD-L1/PD-1 axis and establish a basis for the rational design of combination therapy with immune modulators and radiotherapy.
in vivo TNFα neutralization
Maltby, S., et al. (2014). "Production and differentiation of myeloid cells driven by proinflammatory cytokines in response to acute pneumovirus infection in mice" J Immunol 193(8): 4072-4082. PubMed
Respiratory virus infections are often pathogenic, driving severe inflammatory responses. Most research has focused on localized effects of virus infection and inflammation. However, infection can induce broad-reaching, systemic changes that are only beginning to be characterized. In this study, we assessed the impact of acute pneumovirus infection in C57BL/6 mice on bone marrow hematopoiesis. We hypothesized that inflammatory cytokine production in the lung upregulates myeloid cell production in response to infection. We demonstrate a dramatic increase in the percentages of circulating myeloid cells, which is associated with pronounced elevations in inflammatory cytokines in serum (IFN-gamma, IL-6, CCL2), bone (TNF-alpha), and lung tissue (TNF-alpha, IFN-gamma, IL-6, CCL2, CCL3, G-CSF, osteopontin). Increased hematopoietic stem/progenitor cell percentages (Lineage(-)Sca-I(+)c-kit(+)) were also detected in the bone marrow. This increase was accompanied by an increase in the proportions of committed myeloid progenitors, as determined by colony-forming unit assays. However, no functional changes in hematopoietic stem cells occurred, as assessed by competitive bone marrow reconstitution. Systemic administration of neutralizing Abs to either TNF-alpha or IFN-gamma blocked expansion of myeloid progenitors in the bone marrow and also limited virus clearance from the lung. These findings suggest that acute inflammatory cytokines drive production and differentiation of myeloid cells in the bone marrow by inducing differentiation of committed myeloid progenitors. Our findings provide insight into the mechanisms via which innate immune responses regulate myeloid cell progenitor numbers in response to acute respiratory virus infection.
in vitro TNFα neutralization
Beug, S. T., et al. (2014). "Smac mimetics and innate immune stimuli synergize to promote tumor death" Nat Biotechnol 32(2): 182-190. PubMed
Smac mimetic compounds (SMC), a class of drugs that sensitize cells to apoptosis by counteracting the activity of inhibitor of apoptosis (IAP) proteins, have proven safe in phase 1 clinical trials in cancer patients. However, because SMCs act by enabling transduction of pro-apoptotic signals, SMC monotherapy may be efficacious only in the subset of patients whose tumors produce large quantities of death-inducing proteins such as inflammatory cytokines. Therefore, we reasoned that SMCs would synergize with agents that stimulate a potent yet safe “cytokine storm.” Here we show that oncolytic viruses and adjuvants such as poly(I:C) and CpG induce bystander death of cancer cells treated with SMCs that is mediated by interferon beta (IFN-beta), tumor necrosis factor alpha (TNF-alpha) and/or TNF-related apoptosis-inducing ligand (TRAIL). This combinatorial treatment resulted in tumor regression and extended survival in two mouse models of cancer. As these and other adjuvants have been proven safe in clinical trials, it may be worthwhile to explore their clinical efficacy in combination with SMCs.
in vivo TNFα neutralization
DeBerge, M. P., et al. (2014). "Soluble, but not transmembrane, TNF-alpha is required during influenza infection to limit the magnitude of immune responses and the extent of immunopathology" J Immunol 192(12): 5839-5851. PubMed
TNF-alpha is a pleotropic cytokine that has both proinflammatory and anti-inflammatory functions during influenza infection. TNF-alpha is first expressed as a transmembrane protein that is proteolytically processed to release a soluble form. Transmembrane TNF-alpha (memTNF-alpha) and soluble TNF-alpha (solTNF-alpha) have been shown to exert distinct tissue-protective or tissue-pathologic effects in several disease models. However, the relative contributions of memTNF-alpha or solTNF-alpha in regulating pulmonary immunopathology following influenza infection are unclear. Therefore, we performed intranasal influenza infection in mice exclusively expressing noncleavable memTNF-alpha or lacking TNF-alpha entirely and examined the outcomes. We found that solTNF-alpha, but not memTNF-alpha, was required to limit the size of the immune response and the extent of injury. In the absence of solTNF-alpha, there was a significant increase in the CD8(+) T cell response, including virus-specific CD8(+) T cells, which was due in part to an increased resistance to activation-induced cell death. We found that solTNF-alpha mediates these immunoregulatory effects primarily through TNFR1, because mice deficient in TNFR1, but not TNFR2, exhibited dysregulated immune responses and exacerbated injury similar to that observed in mice lacking solTNF-alpha. We also found that solTNF-alpha expression was required early during infection to regulate the magnitude of the CD8(+) T cell response, indicating that early inflammatory events are critical for the regulation of the effector phase. Taken together, these findings suggest that processing of memTNF-alpha to release solTNF-alpha is a critical event regulating the immune response during influenza infection.
in vivo TNFα neutralization
Kugler, D. G., et al. (2013). "CD4+ T cells are trigger and target of the glucocorticoid response that prevents lethal immunopathology in toxoplasma infection" J Exp Med 210(10): 1919-1927. PubMed
Synthetic glucocorticoids (GCs) are commonly used in the treatment of inflammatory diseases, but the role of endogenous GCs in the regulation of host-protective immune responses is poorly understood. Here we show that GCs are induced during acute Toxoplasma gondii infection and directly control the T cell response to the parasite. When infected with toxoplasma, mice that selectively lack GC receptor (GR) expression in T cells (GR(lck-Cre)) rapidly succumb to infection despite displaying parasite burdens indistinguishable from control animals and unaltered levels of the innate cytokines IL-12 and IL-27. Mortality in the GR(lck-Cre) mice was associated with immunopathology and hyperactive Th1 cell function as revealed by enhanced IFN-gamma and TNF production in vivo. Unexpectedly, these CD4(+) T lymphocytes also overexpressed IL-10. Importantly, CD4(+) T cell depletion in wild-type or GR(lck-Cre) mice led to ablation of the GC response to infection. Moreover, in toxoplasma-infected RAG(-/-) animals, adoptive transfer of CD4(+) T lymphocytes was required for GC induction. These findings establish a novel IL-10-independent immunomodulatory circuit in which CD4(+) T cells trigger a GC response that in turn dampens their own effector function. In the case of T. gondii infection, this self-regulatory pathway is critical for preventing collateral tissue damage and promoting host survival.
in vivo TNFα neutralization
Dietze, K. K., et al. (2013). "Combining regulatory T cell depletion and inhibitory receptor blockade improves reactivation of exhausted virus-specific CD8+ T cells and efficiently reduces chronic retroviral loads" PLoS Pathog 9(12): e1003798. PubMed
Chronic infections with human viruses, such as HIV and HCV, or mouse viruses, such as LCMV or Friend Virus (FV), result in functional exhaustion of CD8(+) T cells. Two main mechanisms have been described that mediate this exhaustion: expression of inhibitory receptors on CD8(+) T cells and expansion of regulatory T cells (Tregs) that suppress CD8(+) T cell activity. Several studies show that blockage of one of these pathways results in reactivation of CD8(+) T cells and partial reduction in chronic viral loads. Using blocking antibodies against PD-1 ligand and Tim-3 and transgenic mice in which Tregs can be selectively ablated, we compared these two treatment strategies and combined them for the first time in a model of chronic retrovirus infection. Blocking inhibitory receptors was more efficient than transient depletion of Tregs in reactivating exhausted CD8(+) T cells and reducing viral set points. However, a combination therapy was superior to any single treatment and further augmented CD8(+) T cell responses and resulted in a sustained reduction in chronic viral loads. These results demonstrate that Tregs and inhibitory receptors are non-overlapping factors in the maintenance of chronic viral infections and that immunotherapies targeting both pathways may be a promising strategy to treat chronic infectious diseases.
in vivo TNFα neutralization
Weinlich, R., et al. (2013). "Protective roles for caspase-8 and cFLIP in adult homeostasis" Cell Rep 5(2): 340-348. PubMed
Caspase-8 or cellular FLICE-like inhibitor protein (cFLIP) deficiency leads to embryonic lethality in mice due to defects in endothelial tissues. Caspase-8(-/-) and receptor-interacting protein kinase-3 (RIPK3)(-/-), but not cFLIP(-/-) and RIPK3(-/-), double-knockout animals develop normally, indicating that caspase-8 antagonizes the lethal effects of RIPK3 during development. Here, we show that the acute deletion of caspase-8 in the gut of adult mice induces enterocyte death, disruption of tissue homeostasis, and inflammation, resulting in sepsis and mortality. Likewise, acute deletion of caspase-8 in a focal region of the skin induces local keratinocyte death, tissue disruption, and inflammation. Strikingly, RIPK3 ablation rescues both phenotypes. However, acute loss of cFLIP in the skin produces a similar phenotype that is not rescued by RIPK3 ablation. TNF neutralization protects from either acute loss of caspase-8 or cFLIP. These results demonstrate that caspase-8-mediated suppression of RIPK3-induced death is required not only during development but also for adult homeostasis. Furthermore, RIPK3-dependent inflammation is dispensable for the skin phenotype.
in vivo TNFα neutralization
Bradley, L. M., et al. (2012). "Matrix metalloprotease 9 mediates neutrophil migration into the airways in response to influenza virus-induced toll-like receptor signaling" PLoS Pathog 8(4): e1002641. PubMed
The early inflammatory response to influenza virus infection contributes to severe lung disease and continues to pose a serious threat to human health. The mechanisms by which neutrophils gain entry to the respiratory tract and their role during pathogenesis remain unclear. Here, we report that neutrophils significantly contributed to morbidity in a pathological mouse model of influenza virus infection. Using extensive immunohistochemistry, bone marrow transfers, and depletion studies, we identified neutrophils as the predominant pulmonary cellular source of the gelatinase matrix metalloprotease (MMP) 9, which is capable of digesting the extracellular matrix. Furthermore, infection of MMP9-deficient mice showed that MMP9 was functionally required for neutrophil migration and control of viral replication in the respiratory tract. Although MMP9 release was toll-like receptor (TLR) signaling-dependent, MyD88-mediated signals in non-hematopoietic cells, rather than neutrophil TLRs themselves, were important for neutrophil migration. These results were extended using multiplex analyses of inflammatory mediators to show that neutrophil chemotactic factor, CCL3, and TNFalpha were reduced in the Myd88(-)/(-) airways. Furthermore, TNFalpha induced MMP9 secretion by neutrophils and blocking TNFalpha in vivo reduced neutrophil recruitment after infection. Innate recognition of influenza virus therefore provides the mechanisms to induce recruitment of neutrophils through chemokines and to enable their motility within the tissue via MMP9-mediated cleavage of the basement membrane. Our results demonstrate a previously unknown contribution of MMP9 to influenza virus pathogenesis by mediating excessive neutrophil migration into the respiratory tract in response to viral replication that could be exploited for therapeutic purposes.
in vivo TNFα neutralization
Quezada, S. A., et al. (2010). "Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts" J Exp Med 207(3): 637-650. PubMed
Adoptive transfer of large numbers of tumor-reactive CD8(+) cytotoxic T lymphocytes (CTLs) expanded and differentiated in vitro has shown promising clinical activity against cancer. However, such protocols are complicated by extensive ex vivo manipulations of tumor-reactive cells and have largely focused on CD8(+) CTLs, with much less emphasis on the role and contribution of CD4(+) T cells. Using a mouse model of advanced melanoma, we found that transfer of small numbers of naive tumor-reactive CD4(+) T cells into lymphopenic recipients induces substantial T cell expansion, differentiation, and regression of large established tumors without the need for in vitro manipulation. Surprisingly, CD4(+) T cells developed cytotoxic activity, and tumor rejection was dependent on class II-restricted recognition of tumors by tumor-reactive CD4(+) T cells. Furthermore, blockade of the coinhibitory receptor CTL-associated antigen 4 (CTLA-4) on the transferred CD4(+) T cells resulted in greater expansion of effector T cells, diminished accumulation of tumor-reactive regulatory T cells, and superior antitumor activity capable of inducing regression of spontaneous mouse melanoma. These findings suggest a novel potential therapeutic role for cytotoxic CD4(+) T cells and CTLA-4 blockade in cancer immunotherapy, and demonstrate the potential advantages of differentiating tumor-reactive CD4(+) cells in vivo over current protocols favoring in vitro expansion and differentiation.