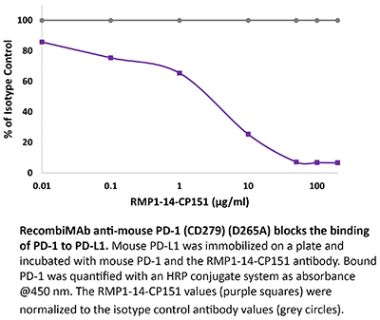
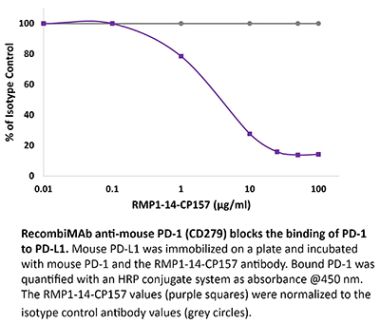
InVivoPlus anti-mouse PD-1 (CD279)

Product Details
The RMP1-14 monoclonal antibody reacts with mouse PD-1 (programmed death-1) also known as CD279. PD-1 is a 50-55 kDa cell surface receptor encoded by the Pdcd1 gene that belongs to the CD28 family of the Ig superfamily. PD-1 is transiently expressed on CD4 and CD8 thymocytes as well as activated T and B lymphocytes and myeloid cells. PD-1 expression declines after successful elimination of antigen. Additionally, Pdcd1 mRNA is expressed in developing B lymphocytes during the pro-B-cell stage. PD-1’s structure includes a ITIM (immunoreceptor tyrosine-based inhibitory motif) suggesting that PD-1 negatively regulates TCR signals. PD-1 signals via binding its two ligands, PD-L1 and PD-L2 both members of the B7 family. Upon ligand binding, PD-1 signaling inhibits T-cell activation, leading to reduced proliferation, cytokine production, and T-cell death. Additionally, PD-1 is known to play key roles in peripheral tolerance and prevention of autoimmune disease in mice as PD-1 knockout animals show dilated cardiomyopathy, splenomegaly, and loss of peripheral tolerance. Induced PD-L1 expression is common in many tumors including squamous cell carcinoma, colon adenocarcinoma, and breast adenocarcinoma. PD-L1 overexpression results in increased resistance of tumor cells to CD8 T cell mediated lysis. In mouse models of melanoma, tumor growth can be transiently arrested via treatment with antibodies which block the interaction between PD-L1 and its receptor PD-1. For these reasons anti-PD-1 mediated immunotherapies are currently being explored as cancer treatments. Like the J43 antibody the RMP1-14 antibody has been shown to block the binding of both mouse PD-L1-Ig and mouse PD-L2-Ig to PD-1.Specifications
| Isotype | Rat IgG2a, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoPlus rat IgG2a isotype control, anti-trinitrophenol |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Syrian Hamster BKH cells transfected with mouse PD-1 cDNA |
| Reported Applications | in vivo blocking of PD-1/PD-L signaling |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin* |
<1EU/mg (<0.001EU/μg) Determined by LAL gel clotting assay |
| Aggregation* |
<5% Determined by SEC |
| Purity |
>95% Determined by SDS-PAGE |
| Sterility | 0.2 μM filtered |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_10949053 |
| Molecular Weight | 150 kDa |
| Murine Pathogen Tests* |
Ectromelia/Mousepox Virus: Negative Hantavirus: Negative K Virus: Negative Lactate Dehydrogenase-Elevating Virus: Negative Lymphocytic Choriomeningitis virus: Negative Mouse Adenovirus: Negative Mouse Cytomegalovirus: Negative Mouse Hepatitis Virus: Negative Mouse Minute Virus: Negative Mouse Norovirus: Negative Mouse Parvovirus: Negative Mouse Rotavirus: Negative Mycoplasma Pulmonis: Negative Pneumonia Virus of Mice: Negative Polyoma Virus: Negative Reovirus Screen: Negative Sendai Virus: Negative Theiler’s Murine Encephalomyelitis: Negative |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
Additional Formats




Recommended Products
-

Recommended Isotype Control(s)
InVivoPlus rat IgG2a isotype control, anti-trinitrophenol
-

Recommended Dilution Buffer
InVivoPure pH 7.0 Dilution Buffer
in vivo blocking of PD-1/PD-L signaling
Triplett, T. A., et al. (2018). "Reversal of indoleamine 2,3-dioxygenase-mediated cancer immune suppression by systemic kynurenine depletion with a therapeutic enzyme" Nat Biotechnol 36(8): 758-764. PubMed
Increased tryptophan (Trp) catabolism in the tumor microenvironment (TME) can mediate immune suppression by upregulation of interferon (IFN)-gamma-inducible indoleamine 2,3-dioxygenase (IDO1) and/or ectopic expression of the predominantly liver-restricted enzyme tryptophan 2,3-dioxygenase (TDO). Whether these effects are due to Trp depletion in the TME or mediated by the accumulation of the IDO1 and/or TDO (hereafter referred to as IDO1/TDO) product kynurenine (Kyn) remains controversial. Here we show that administration of a pharmacologically optimized enzyme (PEGylated kynureninase; hereafter referred to as PEG-KYNase) that degrades Kyn into immunologically inert, nontoxic and readily cleared metabolites inhibits tumor growth. Enzyme treatment was associated with a marked increase in the tumor infiltration and proliferation of polyfunctional CD8(+) lymphocytes. We show that PEG-KYNase administration had substantial therapeutic effects when combined with approved checkpoint inhibitors or with a cancer vaccine for the treatment of large B16-F10 melanoma, 4T1 breast carcinoma or CT26 colon carcinoma tumors. PEG-KYNase mediated prolonged depletion of Kyn in the TME and reversed the modulatory effects of IDO1/TDO upregulation in the TME.
in vivo blocking of PD-1/PD-L signaling
Grasselly, C., et al. (2018). "The Antitumor Activity of Combinations of Cytotoxic Chemotherapy and Immune Checkpoint Inhibitors Is Model-Dependent" Front Immunol 9: 2100. PubMed
In spite of impressive response rates in multiple cancer types, immune checkpoint inhibitors (ICIs) are active in only a minority of patients. Alternative strategies currently aim to combine immunotherapies with conventional agents such as cytotoxic chemotherapies. Here, we performed a study of PD-1 or PDL-1 blockade in combination with reference chemotherapies in four fully immunocompetent mouse models of cancer. We analyzed both the in vivo antitumor response, and the tumor immune infiltrate 4 days after the first treatment. in vivo tumor growth experiments revealed variable responsiveness to ICIs between models. We observed enhanced antitumor effects of the combination of immunotherapy with chemotherapy in the MC38 colon and MB49 bladder models, a lack of response in the 4T1 breast model, and an inhibition of ICIs activity in the MBT-2 bladder model. Flow cytometry analysis of tumor samples showed significant differences in all models between untreated and treated mice. At baseline, all the tumor models studied were predominantly infiltrated with cells harboring an immunosuppressive phenotype. Early alterations of the tumor immune infiltrate after treatment were found to be highly variable. We found that the balance between effector cells and immunosuppressive cells in the tumor microenvironment could be altered with some treatment combinations, but this effect was not always correlated with an impact on in vivo tumor growth. These results show that the combination of cytotoxic chemotherapy with ICIs may result in enhanced, similar or reduced antitumor activity, in a model- and regimen-dependent fashion. The present investigations should help to select appropriate combination regimens for ICIs.
in vivo blocking of PD-1/PD-L signaling
Moynihan, K. D., et al. (2016). "Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses" Nat Med. doi : 10.1038/nm.4200. PubMed
Checkpoint blockade with antibodies specific for cytotoxic T lymphocyte-associated protein (CTLA)-4 or programmed cell death 1 (PDCD1; also known as PD-1) elicits durable tumor regression in metastatic cancer, but these dramatic responses are confined to a minority of patients. This suboptimal outcome is probably due in part to the complex network of immunosuppressive pathways present in advanced tumors, which are unlikely to be overcome by intervention at a single signaling checkpoint. Here we describe a combination immunotherapy that recruits a variety of innate and adaptive immune cells to eliminate large tumor burdens in syngeneic tumor models and a genetically engineered mouse model of melanoma; to our knowledge tumors of this size have not previously been curable by treatments relying on endogenous immunity. Maximal antitumor efficacy required four components: a tumor-antigen-targeting antibody, a recombinant interleukin-2 with an extended half-life, anti-PD-1 and a powerful T cell vaccine. Depletion experiments revealed that CD8+ T cells, cross-presenting dendritic cells and several other innate immune cell subsets were required for tumor regression. Effective treatment induced infiltration of immune cells and production of inflammatory cytokines in the tumor, enhanced antibody-mediated tumor antigen uptake and promoted antigen spreading. These results demonstrate the capacity of an elicited endogenous immune response to destroy large, established tumors and elucidate essential characteristics of combination immunotherapies that are capable of curing a majority of tumors in experimental settings typically viewed as intractable.
in vivo blocking of PD-1/PD-L signaling
Ngiow, S. F., et al. (2015). "A Threshold Level of Intratumor CD8+ T-cell PD1 Expression Dictates Therapeutic Response to Anti-PD1" Cancer Res 75(18): 3800-3811. PubMed
Despite successes, thus far, a significant proportion of the patients treated with anti-PD1 antibodies have failed to respond. We use mouse tumor models of anti-PD1 sensitivity and resistance and flow cytometry to assess tumor-infiltrating immune cells immediately after therapy. We demonstrate that the expression levels of T-cell PD1 (PD1(lo)), myeloid, and T-cell PDL1 (PDL1(hi)) in the tumor microenvironment inversely correlate and dictate the efficacy of anti-PD1 mAb and function of intratumor CD8(+) T cells. In sensitive tumors, we reveal a threshold for PD1 downregulation on tumor-infiltrating CD8(+) T cells below which the release of adaptive immune resistance is achieved. In contrast, PD1(hi) T cells in resistant tumors fail to be rescued by anti-PD1 therapy and remain dysfunctional unless intratumor PDL1(lo) immune cells are targeted. Intratumor Tregs are partly responsible for the development of anti-PD1-resistant tumors and PD1(hi) CD8(+) T cells. Our analyses provide a framework to interrogate intratumor CD8(+) T-cell PD1 and immune PDL1 levels and response in human cancer. Cancer Res; 75(18); 3800-11. (c)2015 AACR.
in vivo blocking of PD-1/PD-L signaling
Evans, E. E., et al. (2015). "Antibody Blockade of Semaphorin 4D Promotes Immune Infiltration into Tumor and Enhances Response to Other Immunomodulatory Therapies" Cancer Immunol Res 3(6): 689-701. PubMed
Semaphorin 4D (SEMA4D, CD100) and its receptor plexin-B1 (PLXNB1) are broadly expressed in murine and human tumors, and their expression has been shown to correlate with invasive disease in several human tumors. SEMA4D normally functions to regulate the motility and differentiation of multiple cell types, including those of the immune, vascular, and nervous systems. In the setting of cancer, SEMA4D-PLXNB1 interactions have been reported to affect vascular stabilization and transactivation of ERBB2, but effects on immune-cell trafficking in the tumor microenvironment (TME) have not been investigated. We describe a novel immunomodulatory function of SEMA4D, whereby strong expression of SEMA4D at the invasive margins of actively growing tumors influences the infiltration and distribution of leukocytes in the TME. Antibody neutralization of SEMA4D disrupts this gradient of expression, enhances recruitment of activated monocytes and lymphocytes into the tumor, and shifts the balance of cells and cytokines toward a proinflammatory and antitumor milieu within the TME. This orchestrated change in the tumor architecture was associated with durable tumor rejection in murine Colon26 and ERBB2(+) mammary carcinoma models. The immunomodulatory activity of anti-SEMA4D antibody can be enhanced by combination with other immunotherapies, including immune checkpoint inhibition and chemotherapy. Strikingly, the combination of anti-SEMA4D antibody with antibody to CTLA-4 acts synergistically to promote complete tumor rejection and survival. Inhibition of SEMA4D represents a novel mechanism and therapeutic strategy to promote functional immune infiltration into the TME and inhibit tumor progression.
in vivo blocking of PD-1/PD-L signaling
Zelenay, S., et al. (2015). "Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity" Cell 162(6): 1257-1270. PubMed
The mechanisms by which melanoma and other cancer cells evade anti-tumor immunity remain incompletely understood. Here, we show that the growth of tumors formed by mutant Braf(V600E) mouse melanoma cells in an immunocompetent host requires their production of prostaglandin E2, which suppresses immunity and fuels tumor-promoting inflammation. Genetic ablation of cyclooxygenases (COX) or prostaglandin E synthases in Braf(V600E) mouse melanoma cells, as well as in Nras(G12D) melanoma or in breast or colorectal cancer cells, renders them susceptible to immune control and provokes a shift in the tumor inflammatory profile toward classic anti-cancer immune pathways. This mouse COX-dependent inflammatory signature is remarkably conserved in human cutaneous melanoma biopsies, arguing for COX activity as a driver of immune suppression across species. Pre-clinical data demonstrate that inhibition of COX synergizes with anti-PD-1 blockade in inducing eradication of tumors, implying that COX inhibitors could be useful adjuvants for immune-based therapies in cancer patients.
in vivo blocking of PD-1/PD-L signaling
Zander, R. A., et al. (2015). "PD-1 Co-inhibitory and OX40 Co-stimulatory Crosstalk Regulates Helper T Cell Differentiation and Anti-Plasmodium Humoral Immunity" Cell Host Microbe 17(5): 628-641. PubMed
The differentiation and protective capacity of Plasmodium-specific T cells are regulated by both positive and negative signals during malaria, but the molecular and cellular details remain poorly defined. Here we show that malaria patients and Plasmodium-infected rodents exhibit atypical expression of the co-stimulatory receptor OX40 on CD4 T cells and that therapeutic enhancement of OX40 signaling enhances helper CD4 T cell activity, humoral immunity, and parasite clearance in rodents. However, these beneficial effects of OX40 signaling are abrogated following coordinate blockade of PD-1 co-inhibitory pathways, which are also upregulated during malaria and associated with elevated parasitemia. Co-administration of biologics blocking PD-1 and promoting OX40 signaling induces excessive interferon-gamma that directly limits helper T cell-mediated support of humoral immunity and decreases parasite control. Our results show that targeting OX40 can enhance Plasmodium control and that crosstalk between co-inhibitory and co-stimulatory pathways in pathogen-specific CD4 T cells can impact pathogen clearance.
in vivo blocking of PD-1/PD-L signaling
Twyman-Saint Victor, C., et al. (2015). "Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer" Nature 520(7547): 373-377. PubMed
Immune checkpoint inhibitors result in impressive clinical responses, but optimal results will require combination with each other and other therapies. This raises fundamental questions about mechanisms of non-redundancy and resistance. Here we report major tumour regressions in a subset of patients with metastatic melanoma treated with an anti-CTLA4 antibody (anti-CTLA4) and radiation, and reproduced this effect in mouse models. Although combined treatment improved responses in irradiated and unirradiated tumours, resistance was common. Unbiased analyses of mice revealed that resistance was due to upregulation of PD-L1 on melanoma cells and associated with T-cell exhaustion. Accordingly, optimal response in melanoma and other cancer types requires radiation, anti-CTLA4 and anti-PD-L1/PD-1. Anti-CTLA4 predominantly inhibits T-regulatory cells (Treg cells), thereby increasing the CD8 T-cell to Treg (CD8/Treg) ratio. Radiation enhances the diversity of the T-cell receptor (TCR) repertoire of intratumoral T cells. Together, anti-CTLA4 promotes expansion of T cells, while radiation shapes the TCR repertoire of the expanded peripheral clones. Addition of PD-L1 blockade reverses T-cell exhaustion to mitigate depression in the CD8/Treg ratio and further encourages oligoclonal T-cell expansion. Similarly to results from mice, patients on our clinical trial with melanoma showing high PD-L1 did not respond to radiation plus anti-CTLA4, demonstrated persistent T-cell exhaustion, and rapidly progressed. Thus, PD-L1 on melanoma cells allows tumours to escape anti-CTLA4-based therapy, and the combination of radiation, anti-CTLA4 and anti-PD-L1 promotes response and immunity through distinct mechanisms.
in vivo blocking of PD-1/PD-L signaling
Vanpouille-Box, C., et al. (2015). "TGFbeta Is a Master Regulator of Radiation Therapy-Induced Antitumor Immunity" Cancer Res 75(11): 2232-2242. PubMed
T cells directed to endogenous tumor antigens are powerful mediators of tumor regression. Recent immunotherapy advances have identified effective interventions to unleash tumor-specific T-cell activity in patients who naturally develop them. Eliciting T-cell responses to a patient’s individual tumor remains a major challenge. Radiation therapy can induce immune responses to model antigens expressed by tumors, but it remains unclear whether it can effectively prime T cells specific for endogenous antigens expressed by poorly immunogenic tumors. We hypothesized that TGFbeta activity is a major obstacle hindering the ability of radiation to generate an in situ tumor vaccine. Here, we show that antibody-mediated TGFbeta neutralization during radiation therapy effectively generates CD8(+) T-cell responses to multiple endogenous tumor antigens in poorly immunogenic mouse carcinomas. Generated T cells were effective at causing regression of irradiated tumors and nonirradiated lung metastases or synchronous tumors (abscopal effect). Gene signatures associated with IFNgamma and immune-mediated rejection were detected in tumors treated with radiation therapy and TGFbeta blockade in combination but not as single agents. Upregulation of programmed death (PD) ligand-1 and -2 in neoplastic and myeloid cells and PD-1 on intratumoral T cells limited tumor rejection, resulting in rapid recurrence. Addition of anti-PD-1 antibodies extended survival achieved with radiation and TGFbeta blockade. Thus, TGFbeta is a fundamental regulator of radiation therapy’s ability to generate an in situ tumor vaccine. The combination of local radiation therapy with TGFbeta neutralization offers a novel individualized strategy for vaccinating patients against their tumors.
in vivo blocking of PD-1/PD-L signaling
Mittal, D., et al. (2014). "Antimetastatic effects of blocking PD-1 and the adenosine A2A receptor" Cancer Res 74(14): 3652-3658. PubMed
Adenosine targeting is an attractive new approach to cancer treatment, but no clinical study has yet examined adenosine inhibition in oncology despite the safe clinical profile of adenosine A2A receptor inhibitors (A2ARi) in Parkinson disease. Metastasis is the main cause of cancer-related deaths worldwide, and therefore we have studied experimental and spontaneous mouse models of melanoma and breast cancer metastasis to demonstrate the efficacy and mechanism of a combination of A2ARi in combination with anti-PD-1 monoclonal antibody (mAb). This combination significantly reduces metastatic burden and prolongs the life of mice compared with either monotherapy alone. Importantly, the combination was only effective when the tumor expressed high levels of CD73, suggesting a tumor biomarker that at a minimum could be used to stratify patients that might receive this combination. The mechanism of the combination therapy was critically dependent on NK cells and IFNgamma, and to a lesser extent, CD8(+) T cells and the effector molecule, perforin. Overall, these results provide a strong rationale to use A2ARi with anti-PD-1 mAb for the treatment of minimal residual and metastatic disease.
in vivo blocking of PD-1/PD-L signaling
McGray, A. J., et al. (2014). "Immunotherapy-induced CD8+ T cells instigate immune suppression in the tumor" Mol Ther 22(1): 206-218. PubMed
Despite clear evidence of immunogenicity, cancer vaccines only provide a modest clinical benefit. To evaluate the mechanisms that limit tumor regression following vaccination, we have investigated the weak efficacy of a highly immunogenic experimental vaccine using a murine melanoma model. We discovered that the tumor adapts rapidly to the immune attack instigated by tumor-specific CD8+ T cells in the first few days following vaccination, resulting in the upregulation of a complex set of biological networks, including multiple immunosuppressive processes. This rapid adaptation acts to prevent sustained local immune attack, despite continued infiltration by increasing numbers of tumor-specific T cells. Combining vaccination with adoptive transfer of tumor-specific T cells produced complete regression of the treated tumors but did not prevent the adaptive immunosuppression. In fact, the adaptive immunosuppressive pathways were more highly induced in regressing tumors, commensurate with the enhanced level of immune attack. Examination of tumor infiltrating T-cell functionality revealed that the adaptive immunosuppression leads to a progressive loss in T-cell function, even in tumors that are regressing. These novel observations that T cells produced by therapeutic intervention can instigate a rapid adaptive immunosuppressive response within the tumor have important implications for clinical implementation of immunotherapies.
in vivo blocking of PD-1/PD-L signaling
John, L. B., et al. (2013). "Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells" Clin Cancer Res 19(20): 5636-5646. PubMed
PURPOSE: To determine the antitumor efficacy and toxicity of a novel combination approach involving adoptive T-cell immunotherapy using chimeric antigen receptor (CAR) T cells with an immunomodulatory reagent for blocking immunosuppression. EXPERIMENTAL DESIGN: We examined whether administration of a PD-1 blocking antibody could increase the therapeutic activity of CAR T cells against two different Her-2(+) tumors. The use of a self-antigen mouse model enabled investigation into the efficacy, mechanism, and toxicity of this combination approach. RESULTS: In this study, we first showed a significant increase in the level of PD-1 expressed on transduced anti-Her-2 CD8(+) T cells following antigen-specific stimulation with PD-L1(+) tumor cells and that markers of activation and proliferation were increased in anti-Her-2 T cells in the presence of anti-PD-1 antibody. In adoptive transfer studies in Her-2 transgenic recipient mice, we showed a significant improvement in growth inhibition of two different Her-2(+) tumors treated with anti-Her-2 T cells in combination with anti-PD-1 antibody. The therapeutic effects observed correlated with increased function of anti-Her-2 T cells following PD-1 blockade. Strikingly, a significant decrease in the percentage of Gr1(+) CD11b(+) myeloid-derived suppressor cells (MDSC) was observed in the tumor microenvironment of mice treated with the combination therapy. Importantly, increased antitumor effects were not associated with any autoimmune pathology in normal tissue expressing Her-2 antigen. CONCLUSION: This study shows that specifically blocking PD-1 immunosuppression can potently enhance CAR T-cell therapy that has significant implications for potentially improving therapeutic outcomes of this approach in patients with cancer.
in vivo blocking of PD-1/PD-L signaling
Holmgaard, R. B., et al. (2013). "Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4" J Exp Med 210(7): 1389-1402. PubMed
The cytotoxic T lymphocyte antigen-4 (CTLA-4)-blocking antibody ipilimumab results in durable responses in metastatic melanoma, though therapeutic benefit has been limited to a fraction of patients. This calls for identification of resistance mechanisms and development of combinatorial strategies. Here, we examine the inhibitory role of indoleamine 2,3-dioxygenase (IDO) on the antitumor efficacy of CTLA-4 blockade. In IDO knockout mice treated with anti-CTLA-4 antibody, we demonstrate a striking delay in B16 melanoma tumor growth and increased overall survival when compared with wild-type mice. This was also observed with antibodies targeting PD-1-PD-L1 and GITR. To highlight the therapeutic relevance of these findings, we show that CTLA-4 blockade strongly synergizes with IDO inhibitors to mediate rejection of both IDO-expressing and nonexpressing poorly immunogenic tumors, emphasizing the importance of the inhibitory role of both tumor- and host-derived IDO. This effect was T cell dependent, leading to enhanced infiltration of tumor-specific effector T cells and a marked increase in the effector-to-regulatory T cell ratios in the tumors. Overall, these data demonstrate the immunosuppressive role of IDO in the context of immunotherapies targeting immune checkpoints and provide a strong incentive to clinically explore combination therapies using IDO inhibitors irrespective of IDO expression by the tumor cells.
in vivo blocking of PD-1/PD-L signaling
van der Werf, N., et al. (2013). "Th2 cell-intrinsic hypo-responsiveness determines susceptibility to helminth infection" PLoS Pathog 9(3): e1003215. PubMed
The suppression of protective Type 2 immunity is a principal factor driving the chronicity of helminth infections, and has been attributed to a range of Th2 cell-extrinsic immune-regulators. However, the intrinsic fate of parasite-specific Th2 cells within a chronic immune down-regulatory environment, and the resultant impact such fate changes may have on host resistance is unknown. We used IL-4gfp reporter mice to demonstrate that during chronic helminth infection with the filarial nematode Litomosoides sigmodontis, CD4(+) Th2 cells are conditioned towards an intrinsically hypo-responsive phenotype, characterised by a loss of functional ability to proliferate and produce the cytokines IL-4, IL-5 and IL-2. Th2 cell hypo-responsiveness was a key element determining susceptibility to L. sigmodontis infection, and could be reversed in vivo by blockade of PD-1 resulting in long-term recovery of Th2 cell functional quality and enhanced resistance. Contrasting with T cell dysfunction in Type 1 settings, the control of Th2 cell hypo-responsiveness by PD-1 was mediated through PD-L2, and not PD-L1. Thus, intrinsic changes in Th2 cell quality leading to a functionally hypo-responsive phenotype play a key role in determining susceptibility to filarial infection, and the therapeutic manipulation of Th2 cell-intrinsic quality provides a potential avenue for promoting resistance to helminths.
in vivo blocking of PD-1/PD-L signaling
Curran, M. A., et al. (2010). "PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors" Proc Natl Acad Sci U S A 107(9): 4275-4280. PubMed
Vaccination with irradiated B16 melanoma cells expressing either GM-CSF (Gvax) or Flt3-ligand (Fvax) combined with antibody blockade of the negative T-cell costimulatory receptor cytotoxic T-lymphocyte antigen-4 (CTLA-4) promotes rejection of preimplanted tumors. Despite CTLA-4 blockade, T-cell proliferation and cytokine production can be inhibited by the interaction of programmed death-1 (PD-1) with its ligands PD-L1 and PD-L2 or by the interaction of PD-L1 with B7-1. Here, we show that the combination of CTLA-4 and PD-1 blockade is more than twice as effective as either alone in promoting the rejection of B16 melanomas in conjunction with Fvax. Adding alphaPD-L1 to this regimen results in rejection of 65% of preimplanted tumors vs. 10% with CTLA-4 blockade alone. Combination PD-1 and CTLA-4 blockade increases effector T-cell (Teff) infiltration, resulting in highly advantageous Teff-to-regulatory T-cell ratios with the tumor. The fraction of tumor-infiltrating Teffs expressing CTLA-4 and PD-1 increases, reflecting the proliferation and accumulation of cells that would otherwise be anergized. Combination blockade also synergistically increases Teff-to-myeloid-derived suppressor cell ratios within B16 melanomas. IFN-gamma production increases in both the tumor and vaccine draining lymph nodes, as does the frequency of IFN-gamma/TNF-alpha double-producing CD8(+) T cells within the tumor. These results suggest that combination blockade of the PD-1/PD-L1- and CTLA-4-negative costimulatory pathways allows tumor-specific T cells that would otherwise be inactivated to continue to expand and carry out effector functions, thereby shifting the tumor microenvironment from suppressive to inflammatory.