InVivoPlus anti-mouse CD8α
Product Details
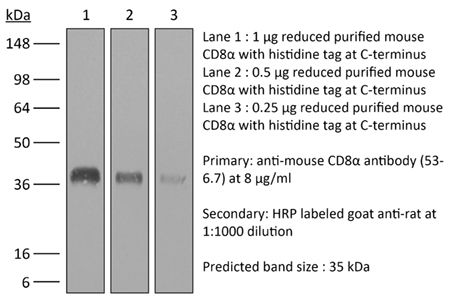
The 53-6.7 monoclonal antibody reacts with mouse CD8α. The CD8 antigen is a transmembrane glycoprotein that acts as a co-receptor for the T cell receptor (TCR). Like the TCR, CD8 binds to class I MHC molecules displayed by antigen presenting cells (APC). CD8 is primarily expressed on the surface of cytotoxic T cells, but can also be found on thymocytes, natural killer cells, and some dendritic cell subsets. CD8 most commonly exists as a heterodimer composed of one CD8α and one CD8β chain however, it can also exist as a homodimer composed of two CD8α chains. Both the CD8α and CD8β chains share significant homology to immunoglobulin variable light chains. The molecular weight of each CD8 chain is approximately 34 kDa. The 53-6.7 antibody exhibits depleting activity when used in vivo.Specifications
| Isotype | Rat IgG2a, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoPlus rat IgG2a isotype control, anti-trinitrophenol |
| Recommended Dilution Buffer | InVivoPure pH 6.5 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Mouse Spleen Cells or Thymocyte Membranes |
| Reported Applications |
in vivo CD8+ T cell depletion Immunofluorescence Flow cytometry Western blot |
| Formulation |
PBS, pH 6.5 Contains no stabilizers or preservatives |
| Endotoxin* |
<1EU/mg (<0.001EU/μg) Determined by LAL gel clotting assay |
| Aggregation* |
<5% Determined by SEC |
| Purity |
>95% Determined by SDS-PAGE |
| Sterility | 0.2 μM filtered |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_1107671 |
| Molecular Weight | 150 kDa |
| Murine Pathogen Tests* |
Ectromelia/Mousepox Virus: Negative Hantavirus: Negative K Virus: Negative Lactate Dehydrogenase-Elevating Virus: Negative Lymphocytic Choriomeningitis virus: Negative Mouse Adenovirus: Negative Mouse Cytomegalovirus: Negative Mouse Hepatitis Virus: Negative Mouse Minute Virus: Negative Mouse Norovirus: Negative Mouse Parvovirus: Negative Mouse Rotavirus: Negative Mycoplasma Pulmonis: Negative Pneumonia Virus of Mice: Negative Polyoma Virus: Negative Reovirus Screen: Negative Sendai Virus: Negative Theiler’s Murine Encephalomyelitis: Negative |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
Additional Formats

Recommended Products
-

Recommended Isotype Control(s)
InVivoPlus rat IgG2a isotype control, anti-trinitrophenol
-

Recommended Dilution Buffer
InVivoPure pH 6.5 Dilution Buffer
in vivo CD8+ T cell depletion, Flow Cytometry
Wang, W., et al. (2018). "RIP1 Kinase Drives Macrophage-Mediated Adaptive Immune Tolerance in Pancreatic Cancer" Cancer Cell 34(5): 757-774 e757. PubMed
Pancreatic ductal adenocarcinoma (PDA) is characterized by immune tolerance and immunotherapeutic resistance. We discovered upregulation of receptor-interacting serine/threonine protein kinase 1 (RIP1) in tumor-associated macrophages (TAMs) in PDA. To study its role in oncogenic progression, we developed a selective small-molecule RIP1 inhibitor with high in vivo exposure. Targeting RIP1 reprogrammed TAMs toward an MHCII(hi)TNFalpha(+)IFNgamma(+) immunogenic phenotype in a STAT1-dependent manner. RIP1 inhibition in TAMs resulted in cytotoxic T cell activation and T helper cell differentiation toward a mixed Th1/Th17 phenotype, leading to tumor immunity in mice and in organotypic models of human PDA. Targeting RIP1 synergized with PD1-and inducible co-stimulator-based immunotherapies. Tumor-promoting effects of RIP1 were independent of its co-association with RIP3. Collectively, our work describes RIP1 as a checkpoint kinase governing tumor immunity.
in vivo CD8+ T cell depletion
Christensen, A. D., et al. (2015). "Depletion of regulatory T cells in a hapten-induced inflammation model results in prolonged and increased inflammation driven by T cells" Clin Exp Immunol 179(3): 485-499. PubMed
Regulatory T cells (Tregs ) are known to play an immunosuppressive role in the response of contact hypersensitivity (CHS), but neither the dynamics of Tregs during the CHS response nor the exaggerated inflammatory response after depletion of Tregs has been characterized in detail. In this study we show that the number of Tregs in the challenged tissue peak at the same time as the ear-swelling reaches its maximum on day 1 after challenge, whereas the number of Tregs in the draining lymph nodes peaks at day 2. As expected, depletion of Tregs by injection of a monoclonal antibody to CD25 prior to sensitization led to a prolonged and sustained inflammatory response which was dependent upon CD8 T cells, and co-stimulatory blockade with cytotoxic T lymphocyte antigen-4-immunoglobulin (CTLA-4-Ig) suppressed the exaggerated inflammation. In contrast, blockade of the interleukin (IL)-10-receptor (IL-10R) did not further increase the exaggerated inflammatory response in the Treg -depleted mice. In the absence of Tregs , the response changed from a mainly acute reaction with heavy infiltration of neutrophils to a sustained response with more chronic characteristics (fewer neutrophils and dominated by macrophages). Furthermore, depletion of Tregs enhanced the release of cytokines and chemokines locally in the inflamed ear and augmented serum levels of the systemic inflammatory mediators serum amyloid (SAP) and haptoglobin early in the response.
Immunofluorescence
Finisguerra, V., et al. (2015). "MET is required for the recruitment of anti-tumoural neutrophils" Nature 522(7556): 349-353. PubMed
Mutations or amplification of the MET proto-oncogene are involved in the pathogenesis of several tumours, which rely on the constitutive engagement of this pathway for their growth and survival. However, MET is expressed not only by cancer cells but also by tumour-associated stromal cells, although its precise role in this compartment is not well characterized. Here we show that MET is required for neutrophil chemoattraction and cytotoxicity in response to its ligand hepatocyte growth factor (HGF). Met deletion in mouse neutrophils enhances tumour growth and metastasis. This phenotype correlates with reduced neutrophil infiltration to both the primary tumour and metastatic sites. Similarly, Met is necessary for neutrophil transudation during colitis, skin rash or peritonitis. Mechanistically, Met is induced by tumour-derived tumour necrosis factor (TNF)-alpha or other inflammatory stimuli in both mouse and human neutrophils. This induction is instrumental for neutrophil transmigration across an activated endothelium and for inducible nitric oxide synthase production upon HGF stimulation. Consequently, HGF/MET-dependent nitric oxide release by neutrophils promotes cancer cell killing, which abates tumour growth and metastasis. After systemic administration of a MET kinase inhibitor, we prove that the therapeutic benefit of MET targeting in cancer cells is partly countered by the pro-tumoural effect arising from MET blockade in neutrophils. Our work identifies an unprecedented role of MET in neutrophils, suggests a potential ‘Achilles’ heel’ of MET-targeted therapies in cancer, and supports the rationale for evaluating anti-MET drugs in certain inflammatory diseases.
in vivo CD8+ T cell depletion
Yamada, D. H., et al. (2015). "Suppression of Fcgamma-receptor-mediated antibody effector function during persistent viral infection" Immunity 42(2): 379-390. PubMed
Understanding how viruses subvert host immunity and persist is essential for developing strategies to eliminate infection. T cell exhaustion during chronic viral infection is well described, but effects on antibody-mediated effector activity are unclear. Herein, we show that increased amounts of immune complexes generated in mice persistently infected with lymphocytic choriomeningitis virus (LCMV) suppressed multiple Fcgamma-receptor (FcgammaR) functions. The high amounts of immune complexes suppressed antibody-mediated cell depletion, therapeutic antibody-killing of LCMV infected cells and human CD20-expressing tumors, as well as reduced immune complex-mediated cross-presentation to T cells. Suppression of FcgammaR activity was not due to inhibitory FcgammaRs or high concentrations of free antibody, and proper FcgammaR functions were restored when persistently infected mice specifically lacked immune complexes. Thus, we identify a mechanism of immunosuppression during viral persistence with implications for understanding effective antibody activity aimed at pathogen control.
in vivo CD8+ T cell depletion, Flow Cytometry
Walsh, K. B., et al. (2014). "Animal model of respiratory syncytial virus: CD8+ T cells cause a cytokine storm that is chemically tractable by sphingosine-1-phosphate 1 receptor agonist therapy" J Virol 88(11): 6281-6293. PubMed
The cytokine storm is an intensified, dysregulated, tissue-injurious inflammatory response driven by cytokine and immune cell components. The cytokine storm during influenza virus infection, whereby the amplified innate immune response is primarily responsible for pulmonary damage, has been well characterized. Now we describe a novel event where virus-specific T cells induce a cytokine storm. The paramyxovirus pneumonia virus of mice (PVM) is a model of human respiratory syncytial virus (hRSV). Unexpectedly, when C57BL/6 mice were infected with PVM, the innate inflammatory response was undetectable until day 5 postinfection, at which time CD8(+) T cells infiltrated into the lung, initiating a cytokine storm by their production of gamma interferon (IFN-gamma) and tumor necrosis factor alpha (TNF-alpha). Administration of an immunomodulatory sphingosine-1-phosphate (S1P) receptor 1 (S1P1R) agonist significantly inhibited PVM-elicited cytokine storm by blunting the PVM-specific CD8(+) T cell response, resulting in diminished pulmonary disease and enhanced survival. IMPORTANCE: A dysregulated overly exuberant immune response, termed a “cytokine storm,” accompanies virus-induced acute respiratory diseases (VARV), is primarily responsible for the accompanying high morbidity and mortality, and can be controlled therapeutically in influenza virus infection of mice and ferrets by administration of sphingosine-1-phosphate 1 receptor (S1P1R) agonists. Here, two novel findings are recorded. First, in contrast to influenza infection, where the cytokine storm is initiated early by the innate immune system, for pneumonia virus of mice (PVM), a model of RSV, the cytokine storm is initiated late in infection by the adaptive immune response: specifically, by virus-specific CD8 T cells via their release of IFN-gamma and TNF-alpha. Blockading these cytokines with neutralizing antibodies blunts the cytokine storm and protects the host. Second, PVM infection is controlled by administration of an S1P1R agonist.
in vivo CD8+ T cell depletion, Flow Cytometry
Uddin, M. N., et al. (2014). "TNF-alpha-dependent hematopoiesis following Bcl11b deletion in T cells restricts metastatic melanoma" J Immunol 192(4): 1946-1953. PubMed
Using several tumor models, we demonstrate that mice deficient in Bcl11b in T cells, although having reduced numbers of T cells in the peripheral lymphoid organs, developed significantly less tumors compared with wild-type mice. Bcl11b(-/-) CD4(+) T cells, with elevated TNF-alpha levels, but not the Bcl11b(-/-) CD8(+) T cells, were required for the reduced tumor burden, as were NK1.1(+) cells, found in increased numbers in Bcl11b(F/F)/CD4-Cre mice. Among NK1.1(+) cells, the NK cell population was predominant in number and was the only population displaying elevated granzyme B levels and increased degranulation, although not increased proliferation. Although the number of myeloid-derived suppressor cells was increased in the lungs with metastatic tumors of Bcl11b(F/F)/CD4-Cre mice, their arginase-1 levels were severely reduced. The increase in NK cell and myeloid-derived suppressor cell numbers was associated with increased bone marrow and splenic hematopoiesis. Finally, the reduced tumor burden, increased numbers of NK cells in the lung, and increased hematopoiesis in Bcl11b(F/F)/CD4-Cre mice were all dependent on TNF-alpha. Moreover, TNF-alpha treatment of wild-type mice also reduced the tumor burden and increased hematopoiesis and the numbers and activity of NK cells in the lung. In vitro treatment with TNF-alpha of lineage-negative hematopoietic progenitors increased NK and myeloid differentiation, further supporting a role of TNF-alpha in promoting hematopoiesis. These studies reveal a novel role for TNF-alpha in the antitumor immune response, specifically in stimulating hematopoiesis and increasing the numbers and activity of NK cells.
in vivo CD8+ T cell depletion, Flow Cytometry
Cyktor, J. C., et al. (2013). "Clonal expansions of CD8+ T cells with IL-10 secreting capacity occur during chronic Mycobacterium tuberculosis infection" PLoS One 8(3): e58612. PubMed
The exact role of CD8(+) T cells during Mycobacterium tuberculosis (Mtb) infection has been heavily debated, yet it is generally accepted that CD8(+) T cells contribute to protection against Mtb. In this study, however, we show that the Mtb-susceptible CBA/J mouse strain accumulates large numbers of CD8(+) T cells in the lung as infection progresses, and that these cells display a dysfunctional and immunosuppressive phenotype (PD-1(+), Tim-3(+), CD122(+)). CD8(+) T cell expansions from the lungs of Mtb-infected CBA/J mice were also capable of secreting the immunosuppressive cytokine interleukin-10 (IL-10), although in vivo CD8(+) T cell depletion did not significantly alter Mtb burden. Further analysis revealed that pulmonary CD8(+) T cells from Mtb-infected CBA/J mice were clonally expanded, preferentially expressing T cell receptor (TcR) Vbeta chain 8 (8.2, 8.3) or Vbeta 14. Although Vbeta8(+) CD8(+) T cells were responsible for the majority of IL-10 production, in vivo depletion of Vbeta8(+) did not significantly change the outcome of Mtb infection, which we hypothesize was a consequence of their dual IL-10/IFN-gamma secreting profiles. Our data demonstrate that IL-10-secreting CD8(+) T cells can arise during chronic Mtb infection, although the significance of this T cell population in tuberculosis pathogenesis remains unclear.
in vivo CD8+ T cell depletion
Hervieu, A., et al. (2013). "Dacarbazine-mediated upregulation of NKG2D ligands on tumor cells activates NK and CD8 T cells and restrains melanoma growth" J Invest Dermatol 133(2): 499-508. PubMed
Dacarbazine (DTIC) is a cytotoxic drug widely used for melanoma treatment. However, the putative contribution of anticancer immune responses in the efficacy of DTIC has not been evaluated. By testing how DTIC affects host immune responses to cancer in a mouse model of melanoma, we unexpectedly found that both natural killer (NK) and CD8(+) T cells were indispensable for DTIC therapeutic effect. Although DTIC did not directly affect immune cells, it triggered the upregulation of NKG2D ligands on tumor cells, leading to NK cell activation and IFNgamma secretion in mice and humans. NK cell-derived IFNgamma subsequently favored upregulation of major histocompatibility complex class I molecules on tumor cells, rendering them sensitive to cytotoxic CD8(+) T cells. Accordingly, DTIC markedly enhanced cytotoxic T lymphocyte antigen 4 inhibition efficacy in vivo in an NK-dependent manner. These results underscore the immunogenic properties of DTIC and provide a rationale to combine DTIC with immunotherapeutic agents that relieve immunosuppression in vivo.
Immunofluorescence
Schwager, K., et al. (2013). "The immunocytokine L19-IL2 eradicates cancer when used in combination with CTLA-4 blockade or with L19-TNF" J Invest Dermatol 133(3): 751-758. PubMed
Systemic high-dose IL2 promotes long-term survival in a subset of metastatic melanoma patients, but this treatment is accompanied by severe toxicities. The immunocytokine L19-IL2, in which IL2 is fused to the human L19 antibody capable of selective accumulation on tumor neovasculature, has recently shown encouraging clinical activity in patients with metastatic melanoma. In this study, we have investigated the therapeutic performance of L19-IL2, administered systemically in combination with a murine anti-CTLA-4 antibody or with a second clinical-stage immunocytokine (L19-TNF) in two syngeneic immunocompetent mouse models of cancer. We observed complete tumor eradications when L19-IL2 was used in combination with CTLA-4 blockade. Interestingly, mice cured from F9 tumors developed new lesions when rechallenged with tumor cells after therapy, whereas mice cured from CT26 tumors were resistant to tumor rechallenge. Similarly, L19-IL2 induced complete remissions when administered in a single intratumoral injection in combination with L19-TNF, whereas the two components did not lead to cures when administered as single agents. These findings provide a rationale for combination trials in melanoma, as the individual therapeutic agents have been extensively studied in clinical trials, and the antigen recognized by the L19 antibody has an identical sequence in mouse and man.
in vivo CD8+ T cell depletion, Flow Cytometry
Hafalla, J. C., et al. (2012). "The CTLA-4 and PD-1/PD-L1 inhibitory pathways independently regulate host resistance to Plasmodium-induced acute immune pathology" PLoS Pathog 8(2): e1002504. PubMed
The balance between pro-inflammatory and regulatory immune responses in determining optimal T cell activation is vital for the successful resolution of microbial infections. This balance is maintained in part by the negative regulators of T cell activation, CTLA-4 and PD-1/PD-L, which dampen effector responses during chronic infections. However, their role in acute infections, such as malaria, remains less clear. In this study, we determined the contribution of CTLA-4 and PD-1/PD-L to the regulation of T cell responses during Plasmodium berghei ANKA (PbA)-induced experimental cerebral malaria (ECM) in susceptible (C57BL/6) and resistant (BALB/c) mice. We found that the expression of CTLA-4 and PD-1 on T cells correlates with the extent of pro-inflammatory responses induced during PbA infection, being higher in C57BL/6 than in BALB/c mice. Thus, ECM develops despite high levels of expression of these inhibitory receptors. However, antibody-mediated blockade of either the CTLA-4 or PD-1/PD-L1, but not the PD-1/PD-L2, pathways during PbA-infection in ECM-resistant BALB/c mice resulted in higher levels of T cell activation, enhanced IFN-gamma production, increased intravascular arrest of both parasitised erythrocytes and CD8(+) T cells to the brain, and augmented incidence of ECM. Thus, in ECM-resistant BALB/c mice, CTLA-4 and PD-1/PD-L1 represent essential, independent and non-redundant pathways for maintaining T cell homeostasis during a virulent malaria infection. Moreover, neutralisation of IFN-gamma or depletion of CD8(+) T cells during PbA infection was shown to reverse the pathologic effects of regulatory pathway blockade, highlighting that the aetiology of ECM in the BALB/c mice is similar to that in C57BL/6 mice. In summary, our results underscore the differential and complex regulation that governs immune responses to malaria parasites.
in vivo CD8+ T cell depletion
Chyou, S., et al. (2011). "Coordinated regulation of lymph node vascular-stromal growth first by CD11c+ cells and then by T and B cells" J Immunol 187(11): 5558-5567. PubMed
Lymph node blood vessels play important roles in the support and trafficking of immune cells. The blood vasculature is a component of the vascular-stromal compartment that also includes the lymphatic vasculature and fibroblastic reticular cells (FRCs). During immune responses as lymph nodes swell, the blood vasculature undergoes a rapid proliferative growth that is initially dependent on CD11c(+) cells and vascular endothelial growth factor (VEGF) but is independent of lymphocytes. The lymphatic vasculature grows with similar kinetics and VEGF dependence, suggesting coregulation of blood and lymphatic vascular growth, but lymphatic growth has been shown to be B cell dependent. In this article, we show that blood vascular, lymphatic, and FRC growth are coordinately regulated and identify two distinct phases of vascular-stromal growth–an initiation phase, characterized by upregulated vascular-stromal proliferation, and a subsequent expansion phase. The initiation phase is CD11c(+) cell dependent and T/B cell independent, whereas the expansion phase is dependent on B and T cells together. Using CCR7(-/-) mice and selective depletion of migratory skin dendritic cells, we show that endogenous skin-derived dendritic cells are not important during the initiation phase and uncover a modest regulatory role for CCR7. Finally, we show that FRC VEGF expression is upregulated during initiation and that dendritic cells can stimulate increased fibroblastic VEGF, suggesting the scenario that lymph node-resident CD11c(+) cells orchestrate the initiation of blood and lymphatic vascular growth in part by stimulating FRCs to upregulate VEGF. These results illustrate how the lymph node microenvironment is shaped by the cells it supports.
in vivo CD8+ T cell depletion
Kumar, D., et al. (2011). "Intranasal administration of an inactivated Yersinia pestis vaccine with interleukin-12 generates protective immunity against pneumonic plague" Clin Vaccine Immunol 18(11): 1925-1935. PubMed
Inhalation of Yersinia pestis causes pneumonic plague, which rapidly progresses to death. A previously licensed killed whole-cell vaccine is presently unavailable due to its reactogenicity and inconclusive evidence of efficacy. The present study now shows that vaccination intranasally (i.n.) with inactivated Y. pestis CO92 (iYp) adjuvanted with interleukin-12 (IL-12) followed by an i.n. challenge with a lethal dose of Y. pestis CO92 prevented bacterial colonization and protected 100% of mice from pneumonic plague. Survival of the vaccinated mice correlated with levels of systemic and lung antibodies, reduced pulmonary pathology and proinflammatory cytokines, and the presence of lung lymphoid cell aggregates. Protection against pneumonic plague was partially dependent upon Fc receptors and could be transferred to naive mice with immune mouse serum. On the other hand, protection was not dependent upon complement, and following vaccination, depletion of CD4 and/or CD8 T cells before challenge did not affect survival. In summary, the results demonstrate the safety, immunogenicity, and protective efficacy of i.n. administered iYp plus IL-12 in a mouse model of pneumonic plague.
in vivo CD8+ T cell depletion
Simma, O., et al. (2009). "Identification of an indispensable role for tyrosine kinase 2 in CTL-mediated tumor surveillance" Cancer Res 69(1): 203-211. PubMed
We showed previously that Tyk2(-/-) natural killer cells lack the ability to lyse leukemic cells. As a consequence, the animals are leukemia prone. Here, we show that the impaired tumor surveillance extends to T cells. Challenging Tyk2(-/-) mice with EL4 thymoma significantly decreased disease latency. The crucial role of Tyk2 for CTL function was further characterized using the ovalbumin-expressing EG7 cells. Tyk2(-/-) OT-1 mice developed EG7-induced tumors significantly faster compared with wild-type (wt) controls. In vivo assays confirmed the defect in CD8(+) cytotoxicity on Tyk2 deficiency and clearly linked it to type I IFN signaling. An impaired CTL activity was only observed in IFNAR1(-/-) animals but not on IFNgamma or IL12p35 deficiency. Accordingly, EG7-induced tumors grew faster in IFNAR1(-/-) and Tyk2(-/-) but not in IFNgamma(-/-) or IL12p35(-/-) mice. Adoptive transfer experiments defined a key role of Tyk2 in CTL-mediated tumor surveillance. In contrast to wt OT-1 cells, Tyk2(-/-) OT-1 T cells were incapable of controlling EG7-induced tumor growth.