InVivoMAb anti-mouse CD4
Product Details
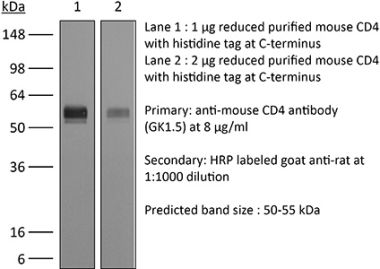
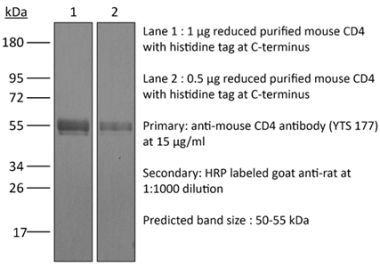
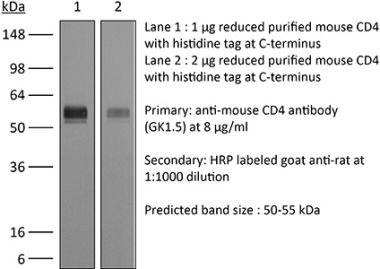
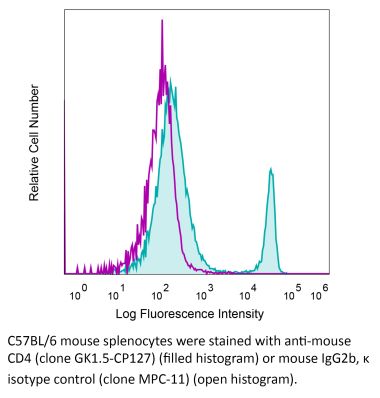
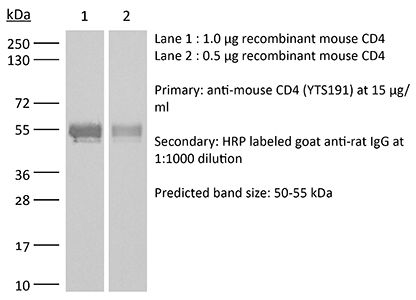
The YTS 191 monoclonal antibody reacts with mouse CD4. The CD4 antigen is a 55 kDa type I cell-surface membrane glycoprotein belonging to the immunoglobulin superfamily. CD4 acts as a co-receptor which in cooperation with the T cell receptor (TCR) interacts with class II MHC molecules displayed by antigen presenting cells (APC). CD4 is expressed by the majority of thymocytes, most helper T cells, a subset of NK-T cells and weakly by dendritic cells and macrophages. CD4 plays an important role in the development of T cells and is required for mature T cells to function optimally. The YTS 191 antibody has been shown to compete with clones GK1.5 and YTS 177 for CD4 binding.Specifications
| Isotype | Rat IgG2b, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb rat IgG2b isotype control, anti-keyhole limpet hemocyanin |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Not available or unknown |
| Reported Applications | in vivo CD4+ T cell depletion |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
<2EU/mg (<0.002EU/μg) Determined by LAL gel clotting assay |
| Purity |
>95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_10950382 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
Recommended Products
-

Recommended Isotype Control(s)
InVivoMAb rat IgG2b isotype control, anti-keyhole limpet hemocyanin
-

Recommended Dilution Buffer
InVivoPure pH 7.0 Dilution Buffer
in vivo CD4+ T cell depletion
Kreiter, S., et al. (2015). "Mutant MHC class II epitopes drive therapeutic immune responses to cancer" Nature 520(7549): 692-696. PubMed
Tumour-specific mutations are ideal targets for cancer immunotherapy as they lack expression in healthy tissues and can potentially be recognized as neo-antigens by the mature T-cell repertoire. Their systematic targeting by vaccine approaches, however, has been hampered by the fact that every patient’s tumour possesses a unique set of mutations (‘the mutanome’) that must first be identified. Recently, we proposed a personalized immunotherapy approach to target the full spectrum of a patient’s individual tumour-specific mutations. Here we show in three independent murine tumour models that a considerable fraction of non-synonymous cancer mutations is immunogenic and that, unexpectedly, the majority of the immunogenic mutanome is recognized by CD4(+) T cells. Vaccination with such CD4(+) immunogenic mutations confers strong antitumour activity. Encouraged by these findings, we established a process by which mutations identified by exome sequencing could be selected as vaccine targets solely through bioinformatic prioritization on the basis of their expression levels and major histocompatibility complex (MHC) class II-binding capacity for rapid production as synthetic poly-neo-epitope messenger RNA vaccines. We show that vaccination with such polytope mRNA vaccines induces potent tumour control and complete rejection of established aggressively growing tumours in mice. Moreover, we demonstrate that CD4(+) T cell neo-epitope vaccination reshapes the tumour microenvironment and induces cytotoxic T lymphocyte responses against an independent immunodominant antigen in mice, indicating orchestration of antigen spread. Finally, we demonstrate an abundance of mutations predicted to bind to MHC class II in human cancers as well by employing the same predictive algorithm on corresponding human cancer types. Thus, the tailored immunotherapy approach introduced here may be regarded as a universally applicable blueprint for comprehensive exploitation of the substantial neo-epitope target repertoire of cancers, enabling the effective targeting of every patient’s tumour with vaccines produced ‘just in time’.
in vivo CD4+ T cell depletion
Burrack, K. S., et al. (2015). "Myeloid Cell Arg1 Inhibits Control of Arthritogenic Alphavirus Infection by Suppressing Antiviral T Cells" PLoS Pathog 11(10): e1005191. PubMed
Arthritogenic alphaviruses, including Ross River virus (RRV) and chikungunya virus (CHIKV), are responsible for explosive epidemics involving millions of cases. These mosquito-transmitted viruses cause inflammation and injury in skeletal muscle and joint tissues that results in debilitating pain. We previously showed that arginase 1 (Arg1) was highly expressed in myeloid cells in the infected and inflamed musculoskeletal tissues of RRV- and CHIKV-infected mice, and specific deletion of Arg1 from myeloid cells resulted in enhanced viral control. Here, we show that Arg1, along with other genes associated with suppressive myeloid cells, is induced in PBMCs isolated from CHIKV-infected patients during the acute phase as well as the chronic phase, and that high Arg1 expression levels were associated with high viral loads and disease severity. Depletion of both CD4 and CD8 T cells from RRV-infected Arg1-deficient mice restored viral loads to levels detected in T cell-depleted wild-type mice. Moreover, Arg1-expressing myeloid cells inhibited virus-specific T cells in the inflamed and infected musculoskeletal tissues, but not lymphoid tissues, following RRV infection in mice, including suppression of interferon-gamma and CD69 expression. Collectively, these data enhance our understanding of the immune response following arthritogenic alphavirus infection and suggest that immunosuppressive myeloid cells may contribute to the duration or severity of these debilitating infections.
in vivo CD4+ T cell depletion
Wensveen, F. M., et al. (2015). "NK cells link obesity-induced adipose stress to inflammation and insulin resistance" Nat Immunol 16(4): 376-385. PubMed
An important cause of obesity-induced insulin resistance is chronic systemic inflammation originating in visceral adipose tissue (VAT). VAT inflammation is associated with the accumulation of proinflammatory macrophages in adipose tissue, but the immunological signals that trigger their accumulation remain unknown. We found that a phenotypically distinct population of tissue-resident natural killer (NK) cells represented a crucial link between obesity-induced adipose stress and VAT inflammation. Obesity drove the upregulation of ligands of the NK cell-activating receptor NCR1 on adipocytes; this stimulated NK cell proliferation and interferon-gamma (IFN-gamma) production, which in turn triggered the differentiation of proinflammatory macrophages and promoted insulin resistance. Deficiency of NK cells, NCR1 or IFN-gamma prevented the accumulation of proinflammatory macrophages in VAT and greatly ameliorated insulin sensitivity. Thus NK cells are key regulators of macrophage polarization and insulin resistance in response to obesity-induced adipocyte stress.
in vivo CD4+ T cell depletion
Yamada, D. H., et al. (2015). "Suppression of Fcgamma-receptor-mediated antibody effector function during persistent viral infection" Immunity 42(2): 379-390. PubMed
Understanding how viruses subvert host immunity and persist is essential for developing strategies to eliminate infection. T cell exhaustion during chronic viral infection is well described, but effects on antibody-mediated effector activity are unclear. Herein, we show that increased amounts of immune complexes generated in mice persistently infected with lymphocytic choriomeningitis virus (LCMV) suppressed multiple Fcgamma-receptor (FcgammaR) functions. The high amounts of immune complexes suppressed antibody-mediated cell depletion, therapeutic antibody-killing of LCMV infected cells and human CD20-expressing tumors, as well as reduced immune complex-mediated cross-presentation to T cells. Suppression of FcgammaR activity was not due to inhibitory FcgammaRs or high concentrations of free antibody, and proper FcgammaR functions were restored when persistently infected mice specifically lacked immune complexes. Thus, we identify a mechanism of immunosuppression during viral persistence with implications for understanding effective antibody activity aimed at pathogen control.
in vivo CD4+ T cell depletion
Steel, C. D., et al. (2014). "Role of peripheral immune response in microglia activation and regulation of brain chemokine and proinflammatory cytokine responses induced during VSV encephalitis" J Neuroimmunol 267(1-2): 50-60. PubMed
We report herein that neuroinvasion by vesicular stomatitis virus (VSV) activates microglia and induces a peripheral dendritic cell (DC)-dependent inflammatory response in the central nervous system (CNS). VSV neuroinvasion rapidly induces multiple brain chemokine and proinflammatory cytokine mRNAs that display bimodal kinetics. Peripheral DC ablation or T cell depletion suppresses the second wave of this response demonstrating that infiltrating T cells are primarily responsible for the bimodal characteristics of this response. The robust infiltrate associated with VSV encephalitis likely depends on sustained production of brain CCL19 and CCR7 expression on infiltrating inflammatory cells.
in vivo CD4+ T cell depletion
Kish, D. D., et al. (2011). "Hapten application to the skin induces an inflammatory program directing hapten-primed effector CD8 T cell interaction with hapten-presenting endothelial cells" J Immunol 186(4): 2117-2126. PubMed
Contact hypersensitivity is a CD8 T cell-mediated response to hapten sensitization and challenge of the skin. Effector CD8 T cell recruitment into the skin parenchyma to elicit the response to hapten challenge requires prior CXCL1/KC-directed neutrophil infiltration within 3-6 h after challenge and is dependent on IFN-gamma and IL-17 produced by the hapten-primed CD8 T cells. Mechanisms directing hapten-primed CD8 T cell localization and activation in the Ag challenge site to induce this early CXCL1 production in response to 2,4-dinitrofluorobenzene were investigated. Both TNF-alpha and IL-17, but not IFN-gamma, mRNA was detectable within 1 h of hapten challenge of sensitized mice and increased thereafter. Expression of ICAM-1 was observed by 1 h after challenge of sensitized and nonsensitized mice and was dependent on TNF-alpha. The induction of IL-17, IFN-gamma, and CXCL1 in the challenge site was not observed when ICAM-1 was absent or neutralized by specific Ab. During the elicitation of the contact hypersensitivity response, endothelial cells expressed ICAM-1 and produced CXCL1 suggesting this as the site of CD8 T cell localization and activation. Endothelial cells isolated from challenged skin of naive and sensitized mice had acquired the hapten and the ability to activate hapten-primed CD8 T cell cytokine production. These results indicate that hapten application to the skin of sensitized animals initiates an inflammatory response promoting hapten-primed CD8 T cell localization to the challenge site through TNF-alpha-induced ICAM-1 expression and CD8 T cell activation to produce IFN-gamma and IL-17 through endothelial cell presentation of hapten.
in vivo CD4+ T cell depletion
Fahey, L. M., et al. (2011). "Viral persistence redirects CD4 T cell differentiation toward T follicular helper cells" J Exp Med 208(5): 987-999. PubMed
CD4 T cell responses are crucial to prevent and control viral infection; however, virus-specific CD4 T cell activity is considered to be rapidly lost during many persistent viral infections. This is largely caused by the fact that during viral persistence CD4 T cells do not produce the classical Th1 cytokines associated with control of acute viral infections. Considering that CD4 T cell help is critical for both CD8 T cell and B cell functions, it is unclear how CD4 T cells can lose responsiveness but continue to sustain long-term control of persistent viral replication. We now demonstrate that CD4 T cell function is not extinguished as a result of viral persistence. Instead, viral persistence and prolonged T cell receptor stimulation progressively redirects CD4 T cell development away from the Th1 response induced during an acute infection toward T follicular helper cells. Importantly, this sustained CD4 T cell functionality is critical to maintain immunity and ultimately aid in the control of persistent viral infection.
in vivo CD4+ T cell depletion
Kish, D. D., et al. (2009). "CD8 T cells producing IL-17 and IFN-gamma initiate the innate immune response required for responses to antigen skin challenge" J Immunol 182(10): 5949-5959. PubMed
Effector CD8 T cell recruitment into the skin in response to Ag challenge requires prior CXCL1/KC-directed neutrophil infiltration. Mechanisms inducing CXCL1 production and the dynamics of neutrophil-CD8 T cell interactions during elicitation of Ag-specific responses in the skin were investigated. CXCL1 and CXCL2/MIP-2 were produced within 3-6 h of Ag challenge at 10-fold higher levels in skin challenge sites of Ag-sensitized vs nonsensitized mice. In the challenge sites of sensitized mice this production decreased at 6-9 h postchallenge to near the levels observed in skin challenge sites of nonsensitized mice but rose to a second peak 12 h after challenge. The elevated early neutrophil chemoattractant production at 3-6 h after skin challenge of sensitized animals required both IFN-gamma and IL-17, produced by distinct populations of Ag-primed CD8 T cells in response to Ag challenge. Although induced by the Ag-primed CD8 T cells, the early CXCL1 and CXCL2 production was accompanied by neutrophil but not CD8 T cell infiltration into the skin Ag challenge site. Infiltration of the CD8 T cells into the challenge site was not observed until 18-24 h after challenge. These results demonstrate an intricate series of early interactions between Ag-specific and innate immune components that regulate the sequential infiltration of neutrophils and then effector T cells into the skin to mediate an immune response.