InVivoMAb anti-human PD-1 (CD279)
Product Details
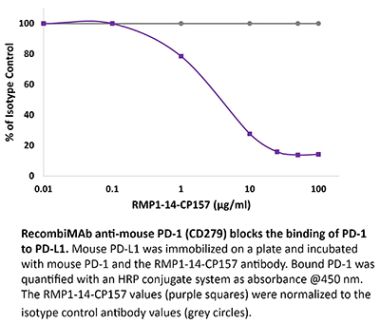
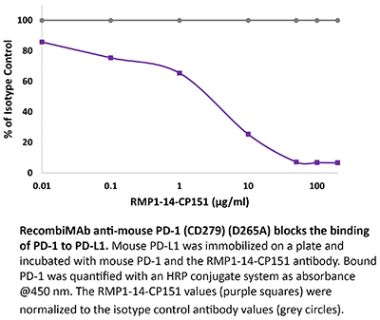
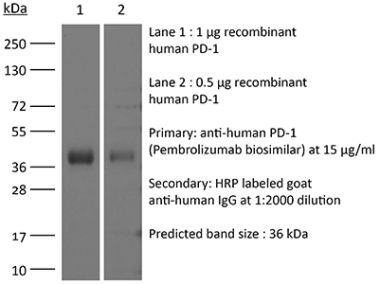
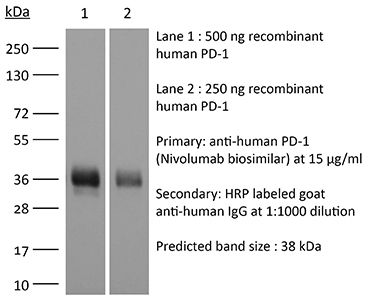
The J116 monoclonal antibody reacts with human PD-1 (programmed death-1) also known as CD279. PD-1 is a 50-55 kDa cell surface receptor encoded by the Pdcd1 gene that belongs to the CD28 family of the Ig superfamily. PD-1 is transiently expressed on CD4 and CD8 thymocytes as well as activated T and B lymphocytes and myeloid cells. PD-1 expression declines after successful elimination of antigen. Additionally, Pdcd1 mRNA is expressed in developing B lymphocytes during the pro-B-cell stage. PD-1’s structure includes a ITIM (immunoreceptor tyrosine-based inhibitory motif) suggesting that PD-1 negatively regulates TCR signals. PD-1 signals via binding its two ligands, PD-L1 and PD-L2 both members of the B7 family. Upon ligand binding, PD-1 signaling inhibits T-cell activation, leading to reduced proliferation, cytokine production, and T cell death. Additionally, PD-1 is known to play key roles in peripheral tolerance and prevention of autoimmune disease in mice as PD-1 knockout animals show dilated cardiomyopathy, splenomegaly, and loss of peripheral tolerance. Induced PD-L1 expression is common in many tumors including squamous cell carcinoma, colon adenocarcinoma, and breast adenocarcinoma. PD-L1 overexpression results in increased resistance of tumor cells to CD8 T cell mediated lysis. In mouse models of melanoma, tumor growth can be transiently arrested via treatment with antibodies which block the interaction between PD-L1 and its receptor PD-1. For these reasons anti-PD-1 mediated immunotherapies are currently being explored as cancer treatments. Binding of the J116 antibody is reported to inhibit PD-1 signal transduction, however, it is not reported to block PD-L1 binding.Specifications
| Isotype | Mouse IgG1, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb mouse IgG1 isotype control, unknown specificity |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Not available or unknown |
| Reported Applications |
in vitro PD-1 neutralization in vivo PD-1 blockade in humanized mice |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
<2EU/mg (<0.002EU/μg) Determined by LAL gel clotting assay |
| Purity |
>95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_10950318 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
Recommended Products
-

Recommended Isotype Control(s)
InVivoMAb mouse IgG1 isotype control, unknown specificity
-

Recommended Dilution Buffer
InVivoPure pH 7.0 Dilution Buffer
in vitro PD-1 neutralization
Tkachev, V., et al. (2015). "Programmed death-1 controls T cell survival by regulating oxidative metabolism" J Immunol 194(12): 5789-5800. PubMed
The coinhibitory receptor programmed death-1 (PD-1) maintains immune homeostasis by negatively regulating T cell function and survival. Blockade of PD-1 increases the severity of graft-versus-host disease (GVHD), but the interplay between PD-1 inhibition and T cell metabolism is not well studied. We found that both murine and human alloreactive T cells concomitantly upregulated PD-1 expression and increased levels of reactive oxygen species (ROS) following allogeneic bone marrow transplantation. This PD-1(Hi)ROS(Hi) phenotype was specific to alloreactive T cells and was not observed in syngeneic T cells during homeostatic proliferation. Blockade of PD-1 signaling decreased both mitochondrial H2O2 and total cellular ROS levels, and PD-1-driven increases in ROS were dependent upon the oxidation of fatty acids, because treatment with etomoxir nullified changes in ROS levels following PD-1 blockade. Downstream of PD-1, elevated ROS levels impaired T cell survival in a process reversed by antioxidants. Furthermore, PD-1-driven changes in ROS were fundamental to establishing a cell’s susceptibility to subsequent metabolic inhibition, because blockade of PD-1 decreased the efficacy of later F1F0-ATP synthase modulation. These data indicate that PD-1 facilitates apoptosis in alloreactive T cells by increasing ROS in a process dependent upon the oxidation of fat. In addition, blockade of PD-1 undermines the potential for subsequent metabolic inhibition, an important consideration given the increasing use of anti-PD-1 therapies in the clinic.
in vivo PD-1 blockade in humanized mice
Tsukahara, T., et al. (2015). "The Tol2 transposon system mediates the genetic engineering of T-cells with CD19-specific chimeric antigen receptors for B-cell malignancies" Gene Ther 22(2): 209-215. PubMed
Engineered T-cell therapy using a CD19-specific chimeric antigen receptor (CD19-CAR) is a promising strategy for the treatment of advanced B-cell malignancies. Gene transfer of CARs to T-cells has widely relied on retroviral vectors, but transposon-based gene transfer has recently emerged as a suitable nonviral method to mediate stable transgene expression. The advantages of transposon vectors compared with viral vectors include their simplicity and cost-effectiveness. We used the Tol2 transposon system to stably transfer CD19-CAR into human T-cells. Normal human peripheral blood lymphocytes were co-nucleofected with the Tol2 transposon donor plasmid carrying CD19-CAR and the transposase expression plasmid and were selectively propagated on NIH3T3 cells expressing human CD19. Expanded CD3(+) T-cells with stable and high-level transgene expression (~95%) produced interferon-gamma upon stimulation with CD19 and specifically lysed Raji cells, a CD19(+) human B-cell lymphoma cell line. Adoptive transfer of these T-cells suppressed tumor progression in Raji tumor-bearing Rag2(-/-)gammac(-/-) immunodeficient mice compared with control mice. These results demonstrate that the Tol2 transposon system could be used to express CD19-CAR in genetically engineered T-cells for the treatment of refractory B-cell malignancies.
in vivo PD-1 blockade in humanized mice
Wang, C., et al. (2013). "Rapamycin-treated human endothelial cells preferentially activate allogeneic regulatory T cells" J Clin Invest 123(4): 1677-1693. PubMed
Human graft endothelial cells (ECs) can act as antigen-presenting cells to initiate allograft rejection by host memory T cells. Rapamycin, an mTOR inhibitor used clinically to suppress T cell responses, also acts on DCs, rendering them tolerogenic. Here, we report the effects of rapamycin on EC alloimmunogenicity. Compared with mock-treated cells, rapamycin-pretreated human ECs (rapa-ECs) stimulated less proliferation and cytokine secretion from allogeneic CD4+ memory cells, an effect mimicked by shRNA knockdown of mTOR or raptor in ECs. The effects of rapamycin persisted for several days and were linked to upregulation of the inhibitory molecules PD-L1 and PD-L2 on rapa-ECs. Additionally, rapa-ECs produced lower levels of the inflammatory cytokine IL-6. CD4+ memory cells activated by allogeneic rapa-ECs became hyporesponsive to restimulation in an alloantigen-specific manner and contained higher percentages of suppressive CD4+CD25(hi)CD127(lo)FoxP3+ cells that did not produce effector cytokines. In a human-mouse chimeric model of allograft rejection, rapamycin pretreatment of human arterial allografts increased graft EC expression of PD-L1 and PD-L2 and reduced subsequent infiltration of allogeneic effector T cells into the artery intima and intimal expansion. Preoperative conditioning of allograft ECs with rapamycin could potentially reduce immune-mediated rejection.
in vitro PD-1 neutralization
Singh, A., et al. (2012). "Foxp3+ regulatory T cells among tuberculosis patients: impact on prognosis and restoration of antigen specific IFN-gamma producing T cells" PLoS One 7(9): e44728. PubMed
CD4(+)CD25(+)Foxp3(+) regulatory T cells (Treg) and programmed death-1 (PD-1) molecules have emerged as pivotal players in immune suppression of chronic diseases. However, their impact on the disease severity, therapeutic response and restoration of immune response in human tuberculosis remains unclear. Here, we describe the possible role of Treg cells, their M. tuberculosis driven expansion and contribution of PD-1 pathway to the suppressive function of Treg cells among pulmonary tuberculosis (PTB) patients. Multicolor flow cytometry, cell culture, cells sorting and ELISA were employed to execute the study. Our results showed significant increase in frequency of antigen-reactive Treg cells, which gradually declined during successful therapy and paralleled with decline of M. tuberculosis-specific IL-10 along with elevation of IFN-gamma production, and raising the IFN-gamma/IL-4 ratio. Interestingly, persistence of Treg cells tightly correlated with MDR tuberculosis. Also, we show that blocking PD-1/PD-L1 pathway abrogates Treg-mediated suppression, suggesting that the PD-1/PD-L1 pathway is required for Treg-mediated suppression of the antigen-specific T cells. Treg cells possibly play a role in dampening the effector immune response and abrogating PD-1 pathway on Treg cells significantly rescued protective T cell response, suggesting its importance in immune restoration among tuberculosis patients.
in vitro PD-1 neutralization
Rosignoli, G., et al. (2009). "Programmed death (PD)-1 molecule and its ligand PD-L1 distribution among memory CD4 and CD8 T cell subsets in human immunodeficiency virus-1-infected individuals" Clin Exp Immunol 157(1): 90-97. PubMed
Human immunodeficiency virus (HIV)-1 causes T cell anergy and affects T cell maturation. Various mechanisms are responsible for impaired anti-HIV-1-specific responses: programmed death (PD)-1 molecule and its ligand PD-L1 are negative regulators of T cell activity and their expression is increased during HIV-1 infection. This study examines correlations between T cell maturation, expression of PD-1 and PD-L1, and the effects of their blockade. Peripheral blood mononuclear cells (PBMC) from 24 HIV-1(+) and 17 uninfected individuals were phenotyped for PD-1 and PD-L1 expression on CD4(+) and CD8(+) T cell subsets. The effect of PD-1 and PD-L1 blockade on proliferation and interferon (IFN)-gamma production was tested on eight HIV-1(+) patients. Naive (CCR7(+)CD45RA(+)) CD8(+) T cells were reduced in HIV-1 aviraemic (P = 0.0065) and viraemic patients (P = 0.0130); CD8 T effector memory subsets [CCR7(-)CD45RA(-)(T(EM))] were increased in HIV-1(+) aviraemic (P = 0.0122) and viraemic (P = 0.0023) individuals versus controls. PD-1 expression was increased in CD4 naive (P = 0.0496), central memory [CCR7(+)CD45RA(-) (T(CM)); P = 0.0116], T(EM) (P = 0.0037) and CD8 naive T cells (P = 0.0133) of aviraemic HIV-1(+) versus controls. PD-L1 was increased in CD4 T(EMRA) (CCR7(-)CD45RA(+), P = 0.0119), CD8 T(EM) (P = 0.0494) and CD8 T(EMRA) (P = 0.0282) of aviraemic HIV-1(+)versus controls. PD-1 blockade increased HIV-1-specific proliferative responses in one of eight patients, whereas PD-L1 blockade restored responses in four of eight patients, but did not increase IFN-gamma-production. Alteration of T cell subsets, accompanied by increased PD-1 and PD-L1 expression in HIV-1 infection contributes to anergy and impaired anti-HIV-1-specific responses which are not rescued when PD-1 is blocked, in contrast to when PD-L1 is blocked, due possibly to an ability to bind to receptors other than PD-1.
in vitro PD-1 neutralization
urado, J. O., et al. (2008). "Programmed death (PD)-1:PD-ligand 1/PD-ligand 2 pathway inhibits T cell effector functions during human tuberculosis" J Immunol 181(1): 116-125. PubMed
Protective immunity against Mycobacterium tuberculosis requires the generation of cell-mediated immunity. We investigated the expression and role of programmed death 1 (PD-1) and its ligands, molecules known to modulate T cell activation, in the regulation of IFN-gamma production and lytic degranulation during human tuberculosis. We demonstrated that specific Ag-stimulation increased CD3+PD-1+ lymphocytes in peripheral blood and pleural fluid from tuberculosis patients in direct correlation with IFN-gamma production from these individuals. Moreover, M. tuberculosis-induced IFN-gamma participated in the up-regulation of PD-1 expression. Blockage of PD-1 or PD-1 and its ligands (PD-Ls: PD-L1, PD-L2) enhanced the specific degranulation of CD8+ T cells and the percentage of specific IFN-gamma-producing lymphocytes against the pathogen, demonstrating that the PD-1:PD-Ls pathway inhibits T cell effector functions during active M. tuberculosis infection. Furthermore, the simultaneous blockage of the inhibitory receptor PD-1 together with the activation of the costimulatory protein signaling lymphocytic activation molecule led to the promotion of protective IFN-gamma responses to M. tuberculosis, even in patients with weak cell-mediated immunity against the bacteria. Together, we demonstrated that PD-1 interferes with T cell effector functions against M. tuberculosis, suggesting that PD-1 has a key regulatory role during the immune response of the host to the pathogen.