InVivoMAb anti-mouse IL-18
Product Details
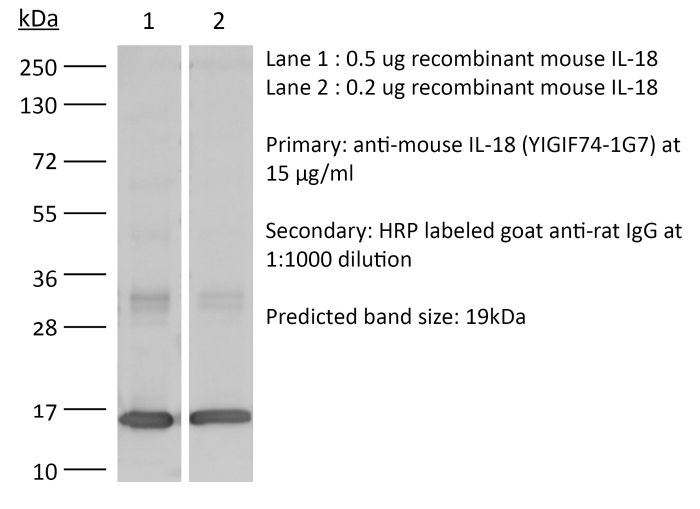
The YIGIF74-1G7 monoclonal antibody reacts with mouse IL-18, an 18 kDa pro-inflammatory cytokine. IL-18 is expressed by activated macrophages, keratinocytes, Kupffer cells, intestinal epithelial cells, and osteoblasts. IL-18 has been shown to activate NF-κB, induce Fas ligand expression, induce both CC and CXC chemokine expression, and enhance the production of IFNγ and GM-CSF.Specifications
| Isotype | Rat IgG2a, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb rat IgG2a isotype control, anti-trinitrophenol |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Not available or unknown |
| Reported Applications |
in vivo IL-18 neutralization in vitro IL-18 neutralization |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
<2EU/mg (<0.002EU/μg) Determined by LAL gel clotting assay |
| Purity |
>95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_2687719 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
Recommended Products
-

Recommended Isotype Control(s)
InVivoMAb rat IgG2a isotype control, anti-trinitrophenol
-

Recommended Dilution Buffer
InVivoPure pH 7.0 Dilution Buffer
in vivo IL-18 neutralization
Cohen, T. S., et al. (2018). "S. aureus Evades Macrophage Killing through NLRP3-Dependent Effects on Mitochondrial Trafficking" Cell Rep 22(9): 2431-2441. PubMed
Clinical severity of Staphylococcus aureus respiratory infection correlates with alpha toxin (AT) expression. AT activates the NLRP3 inflammasome; deletion of Nlrp3, or AT neutralization, protects mice from lethal S. aureus pneumonia. We tested the hypothesis that this protection is not due to a reduction in inflammasome-dependent cytokines (IL-1beta/IL-18) but increased bactericidal function of macrophages. In vivo, neutralization of AT or NLRP3 improved bacterial clearance and survival, while blocking IL-1beta/IL-18 did not. Primary human monocytes were used in vitro to determine the mechanism through which NLRP3 alters bacterial killing. In cells treated with small interfering RNA (siRNA) targeting NLRP3 or infected with AT-null S. aureus, mitochondria co-localize with bacterial-containing phagosomes. Mitochondrial engagement activates caspase-1, a process dependent on complex II of the electron transport chain, near the phagosome, promoting its acidification. These data demonstrate a mechanism utilized by S. aureus to sequester itself from antimicrobial processes within the cell.
in vivo IL-18 neutralization
Robinson, K. M., et al. (2018). "The inflammasome potentiates influenza/Staphylococcus aureus superinfection in mice" JCI Insight 3(7). PubMed
Secondary bacterial respiratory infections are commonly associated with both acute and chronic lung injury. Influenza complicated by bacterial pneumonia is an effective model to study host defense during pulmonary superinfection due to its clinical relevance. Multiprotein inflammasomes are responsible for IL-1β production in response to infection and drive tissue inflammation. In this study, we examined the role of the inflammasome during viral/bacterial superinfection. We demonstrate that ASC-/- mice are protected from bacterial superinfection and produce sufficient quantities of IL-1β through an apoptosis-associated speck-like protein containing CARD (ASC) inflammasome-independent mechanism. Despite the production of IL-1β by ASC-/- mice in response to bacterial superinfection, these mice display decreased lung inflammation. A neutrophil elastase inhibitor blocked ASC inflammasome-independent production of IL-1β and the IL-1 receptor antagonist, anakinra, confirmed that IL-1 remains crucial to the clearance of bacteria during superinfection. Delayed inhibition of NLRP3 during influenza infection by MCC950 decreases bacterial burden during superinfection and leads to decreased inflammatory cytokine production. Collectively, our results demonstrate that ASC augments the clearance of bacteria, but can also contribute to inflammation and mortality. ASC should be considered as a therapeutic target to decrease morbidity and mortality during bacterial superinfection.
in vivo IL-18 neutralization, in vitro IL-18 neutralization
Molgora, M., et al. (2017). "IL-1R8 is a checkpoint in NK cells regulating anti-tumour and anti-viral activity" Nature 551(7678): 110-114. PubMed
Interleukin-1 receptor 8 (IL-1R8, also known as single immunoglobulin IL-1R-related receptor, SIGIRR, or TIR8) is a member of the IL-1 receptor (ILR) family with distinct structural and functional characteristics, acting as a negative regulator of ILR and Toll-like receptor (TLR) downstream signalling pathways and inflammation. Natural killer (NK) cells are innate lymphoid cells which mediate resistance against pathogens and contribute to the activation and orientation of adaptive immune responses. NK cells mediate resistance against haematopoietic neoplasms but are generally considered to play a minor role in solid tumour carcinogenesis. Here we report that IL-1R8 serves as a checkpoint for NK cell maturation and effector function. Its genetic blockade unleashes NK-cell-mediated resistance to hepatic carcinogenesis, haematogenous liver and lung metastasis, and cytomegalovirus infection.
in vivo IL-18 neutralization
Chudnovskiy, A., et al. (2016). "Host-Protozoan Interactions Protect from Mucosal Infections through Activation of the Inflammasome" Cell 167(2): 444-456 e414. PubMed
While conventional pathogenic protists have been extensively studied, there is an underappreciated constitutive protist microbiota that is an integral part of the vertebrate microbiome. The impact of these species on the host and their potential contributions to mucosal immune homeostasis remain poorly studied. Here, we show that the protozoan Tritrichomonas musculis activates the host epithelial inflammasome to induce IL-18 release. Epithelial-derived IL-18 promotes dendritic cell-driven Th1 and Th17 immunity and confers dramatic protection from mucosal bacterial infections. Along with its role as a “protistic” antibiotic, colonization with T. musculis exacerbates the development of T-cell-driven colitis and sporadic colorectal tumors. Our findings demonstrate a novel mutualistic host-protozoan interaction that increases mucosal host defenses at the cost of an increased risk of inflammatory disease.