InVivoMAb anti-rat CD28
Product Details
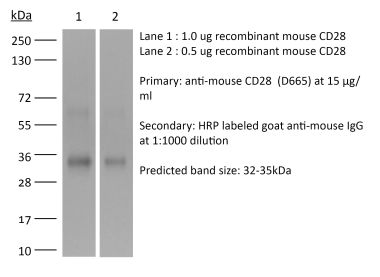
The JJ319 monoclonal antibody reacts with rat CD28, a 45 kDa costimulatory receptor and a member of the Ig superfamily. CD28 is expressed by thymocytes, most peripheral T cells, and NK cells. CD28 is a receptor for CD80 (B7-1) and CD86 (B7-2). Signaling through CD28 augments IL-2 and IL-2 receptor expression as well as cytotoxicity of CD3-activated T cells.Specifications
| Isotype | Mouse IgG1, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb mouse IgG1 isotype control, unknown specificity |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Rat CD28-transfected BALB/c mouse A2OJ cell line |
| Reported Applications |
in vitro T cell stimulation/activation Flow cytometry |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
<2EU/mg (<0.002EU/μg) Determined by LAL gel clotting assay |
| Purity |
>95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_1107626 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
Recommended Products
-

Recommended Isotype Control(s)
InVivoMAb mouse IgG1 isotype control, unknown specificity
-

Recommended Dilution Buffer
InVivoPure pH 7.0 Dilution Buffer
in vitro T cell stimulation/activation
Guendisch, U., et al. (2017). "Pharmacological inhibition of RORγt suppresses the Th17 pathway and alleviates arthritis in vivo" PLoS One 12(11): e0188391. PubMed
Retinoic acid receptor-related-orphan-receptor-C (RORγt) is the key transcription factor that is driving the differentiation of IL-17 producing T-helper 17 (Th17) cells that are implicated in the pathology of various autoimmune and inflammatory diseases. Based on the importance of RORγt in promoting Th17-driven pathology, there is considerable interest to develop low-molecular-weight compounds with the aim of inhibiting the transcriptional activity of this nuclear hormone receptor. In this article, we describe the in vitro and in vivo pharmacology of a potent and selective small-molecular-weight RORγt inverse agonist. The compound binds to the ligand binding domain (LBD) of RORγt leading to displacement of a co-activator peptide. We show for the first time that a RORγt inverse agonist down-regulates permissive histone H3 acetylation and methylation at the IL17A and IL23R promoter regions, thereby providing insight into the transcriptional inhibition of RORγt-dependent genes. Consistent with this, the compound effectively reduced IL-17A production by polarized human T-cells and γδT-cells and attenuated transcription of RORγt target genes. The inhibitor showed good in vivo efficacy in an antigen-induced arthritis model in rats and reduced the frequencies of IL-17A producing cells in ex vivo recall assays. In summary, we demonstrate that inhibiting RORγt by a low-molecular-weight inhibitor results in efficient and selective blockade of the pro-inflammatory Th17/IL-17A pathway making it an attractive target for Th17-mediated disorders.
in vitro T cell stimulation/activation
Guntermann, C., et al. (2017). "Retinoic-acid-orphan-receptor-C inhibition suppresses Th17 cells and induces thymic aberrations" JCI Insight 2(5): e91127. PubMed
Retinoic-acid-orphan-receptor-C (RORC) is a master regulator of Th17 cells, which are pathogenic in several autoimmune diseases. Genetic Rorc deficiency in mice, while preventing autoimmunity, causes early lethality due to metastatic thymic T cell lymphomas. We sought to determine whether pharmacological RORC inhibition could be an effective and safe therapy for autoimmune diseases by evaluating its effects on Th17 cell functions and intrathymic T cell development. RORC inhibitors effectively inhibited Th17 differentiation and IL-17A production, and delayed-type hypersensitivity reactions. In vitro, RORC inhibitors induced apoptosis, as well as Bcl2l1 and BCL2L1 mRNA downregulation, in mouse and nonhuman primate thymocytes, respectively. Chronic, 13-week RORC inhibitor treatment in rats caused progressive thymic alterations in all analyzed rats similar to those in Rorc-deficient mice prior to T cell lymphoma development. One rat developed thymic cortical hyperplasia with preneoplastic features, including increased mitosis and reduced IKAROS expression, albeit without skewed T cell clonality. In summary, pharmacological inhibition of RORC not only blocks Th17 cell development and related cytokine production, but also recapitulates thymic aberrations seen in Rorc-deficient mice. While RORC inhibition may offer an effective therapeutic principle for Th17-mediated diseases, T cell lymphoma with chronic therapy remains an apparent risk.
in vitro T cell stimulation/activation
Pedros, C., et al. (2015). "An Epistatic Interaction between Themis1 and Vav1 Modulates Regulatory T Cell Function and Inflammatory Bowel Disease Development" J Immunol 195(4): 1608-1616. PubMed
The development of inflammatory diseases depends on complex interactions between several genes and various environmental factors. Discovering new genetic risk factors and understanding the mechanisms whereby they influence disease development is of paramount importance. We previously reported that deficiency in Themis1, a new actor of TCR signaling, impairs regulatory T cell (Treg) function and predisposes Brown-Norway (BN) rats to spontaneous inflammatory bowel disease (IBD). In this study, we reveal that the epistasis between Themis1 and Vav1 controls the occurrence of these phenotypes. Indeed, by contrast with BN rats, Themis1 deficiency in Lewis rats neither impairs Treg suppressive functions nor induces pathological manifestations. By using congenic lines on the BN genomic background, we show that the impact of Themis1 deficiency on Treg suppressive functions depends on a 117-kb interval coding for a R63W polymorphism that impacts Vav1 expression and functions. Indeed, the introduction of a 117-kb interval containing the Lewis Vav1-R63 variant restores Treg function and protects Themis1-deficient BN rats from spontaneous IBD development. We further show that Themis1 binds more efficiently to the BN Vav1-W63 variant and is required to stabilize its recruitment to the transmembrane adaptor LAT and to fully promote the activation of Erk kinases. Together, these results highlight the importance of the signaling pathway involving epistasis between Themis1 and Vav1 in the control of Treg suppressive function and susceptibility to IBD development.
in vitro T cell stimulation/activation
Fichtner, A. S., et al. (2015). "Function and expression of CD1d and invariant natural killer T-cell receptor in the cotton rat (Sigmodon hispidus)" Immunology . PubMed
The cotton rat (Sigmodon hispidus) belongs to the rodent family of Cricetidae and provides a powerful model to study the pathogenesis of human respiratory viruses and measles virus. Recent studies in other rodent models have suggested a role for invariant natural killer T (iNKT) cells in antiviral immunity and vaccination against respiratory virus infections. Using new experimental tools, we provide the first evidence for a functional CD1d cell molecule (crCD1d) and iNKT T-cell receptor in cotton rats. The crCD1d cDNA sequence was identified and crCD1d transductants showed that monoclonal antibody WTH-2 stains crCD1d as efficiently as mouse or rat CD1d. The expression of crCD1d was clearly weaker for thymocytes and B cells, and higher for T cells, which is different to what is found in murine species. The antigen-presenting capacity of crCD1d was demonstrated with crCD1d-immunoglobulin dimers loaded with the glycolipid PBS57, which bound iNKT T-cell receptors. Evidence for functional cotton rat iNKT cells was provided by detection of interferon-gamma and interleukin-4 in cultures of splenocytes stimulated with PBS57 and alpha-galactosylceramide and by specific staining of about 0.2% of splenocytes with PBS57-loaded crCD1d dimers. Canonical AV14/AJ18 rearrangements were identified and found to contain multiple members of the AV14 (AV11) family. One of them was expressed and found to bind CD1d dimers. In summary, these data provide the first evidence for functional CD1d molecules and iNKT T-cell receptors in cotton rats and provide the tools to analyse them both in the cotton rat model of infectious diseases.
in vitro T cell stimulation/activation
Chen, X. L., et al. (2015). "GIMAP5 Deficiency Is Associated with Increased AKT Activity in T Lymphocytes" PLoS One 10(10): e0139019. PubMed
Long-term survival of T lymphocytes in quiescent state is essential to maintain their cell numbers in secondary lymphoid organs. In mice and in rats, the loss of functional GTPase of the immune associated nucleotide binding protein 5 (GIMAP5) causes peripheral T lymphopenia due to spontaneous death of T cells. The underlying mechanism responsible for the disruption of quiescence in Gimap5 deficient T cells remains largely unknown. In this study, we show that loss of functional Gimap5 results in increased basal activation of mammalian target of rapamycin (mTOR), independent of protein phosphatase 2A (PP2A) or AMP-activated protein kinase (AMPK). Our results suggest that the constitutive activation of the phosphoinositide 3-kinase (PI3K) pathway may be one of the consequences of the absence of functional GIMAP5.
in vitro T cell stimulation/activation, Flow Cytometry
Picarda, E., et al. (2014). "MHC-derived allopeptide activates TCR-biased CD8+ Tregs and suppresses organ rejection" J Clin Invest 124(6): 2497-2512. PubMed
In a rat heart allograft model, preventing T cell costimulation with CD40Ig leads to indefinite allograft survival, which is mediated by the induction of CD8+CD45RClo regulatory T cells (CD8+CD40Ig Tregs) interacting with plasmacytoid dendritic cells (pDCs). The role of TCR-MHC-peptide interaction in regulating Treg activity remains a topic of debate. Here, we identified a donor MHC class II-derived peptide (Du51) that is recognized by TCR-biased CD8+CD40Ig Tregs and activating CD8+CD40Ig Tregs in both its phenotype and suppression of antidonor alloreactive T cell responses. We generated a labeled tetramer (MHC-I RT1.Aa/Du51) to localize and quantify Du51-specific T cells within rat cardiac allografts and spleen. RT1.Aa/Du51-specific CD8+CD40Ig Tregs were the most suppressive subset of the total Treg population, were essential for in vivo tolerance induction, and expressed a biased, restricted Vbeta11-TCR repertoire in the spleen and the graft. Finally, we demonstrated that treatment of transplant recipients with the Du51 peptide resulted in indefinite prolongation of allograft survival. These results show that CD8+CD40Ig Tregs recognize a dominant donor antigen, resulting in TCR repertoire alterations in the graft and periphery. Furthermore, this allopeptide has strong therapeutic activity and highlights the importance of TCR-peptide-MHC interaction for Treg generation and function.