InVivoMAb anti-mouse VEGFR-2
Product Details
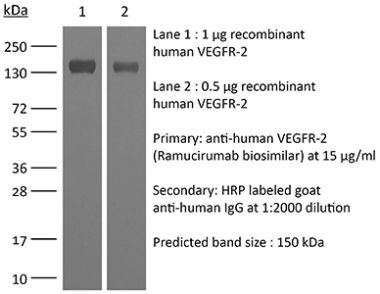
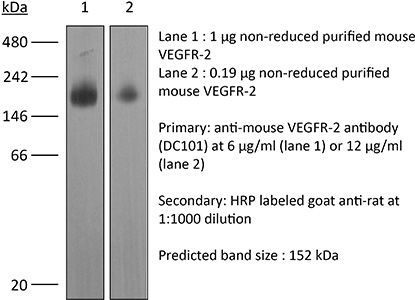
The DC101 monoclonal antibody reacts with mouse VEGFR-2 (vascular endothelial growth factor receptor 2) also known as CD309, KDR, and Flk-1. VEGFR-2 is a member of the tyrosine protein kinase family. Upon binding to its ligand VEGF, VEGFR-2 pays key roles in vascular development and permeability. VEGFR-2 is expressed on endothelial cells at high levels in adult heart, lung, kidney, brain, and skeletal muscle as well as other tissues at lower levels. The DC101 antibody has been shown to inhibit VEGFR-2 signaling in vivo.Specifications
| Isotype | Rat IgG1, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb rat IgG1 isotype control, anti-horseradish peroxidase |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Mouse VEGFR-2-SEAPs soluble receptor |
| Reported Applications |
in vivo blocking of VEGF/VEGFR-2 signaling in vitro blocking of VEGFR signaling Western blot |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
<2EU/mg (<0.002EU/μg) Determined by LAL gel clotting assay |
| Purity |
>95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_1107766 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
Additional Formats


Recommended Products
-

Recommended Isotype Control(s)
InVivoMAb rat IgG1 isotype control, anti-horseradish peroxidase
-

Recommended Dilution Buffer
InVivoPure pH 7.0 Dilution Buffer
in vivo blocking of VEGF/VEGFR-2 signaling
Ding, X., et al. (2015). "Distinct functions of epidermal and myeloid-derived VEGF-A in skin tumorigenesis mediated by HPV8" Cancer Res 75(2): 330-343. PubMed
Beta human papillomaviruses (HPV) have been suspected to be carcinogenic in nonmelanoma skin cancers (NMSC), but the basis for potential viral contributions to these cancers is poorly understood. In particular, it is unresolved how HPV-infected keratinocytes escape cell-cycle control and whether their cross-talk with immune cells is critical for tumorigenesis. In nonviral preclinical models, the angiogenic cytokine VEGF-A has been identified as a critical regulator of NMSC. In this study, we dissected the contribution of epidermal versus myeloid cell-derived VEGF-A in HPV-mediated skin cancer by interbreeding an HPV8 transgenic mouse model with a conditional disruption of VEGF-A restricted to either epidermal or myeloid cells. Although only epidermal-derived VEGF-A was essential for initiation of skin tumor development, both spontaneously and UV-light triggered, both epidermal and myeloid cell-derived VEGF-A contributed to regeneration-induced tumorigenesis upon HPV8 overexpression, partly not only through a paracrine effect on endothelial cells, but also most probably through an additional autocrine effect on epidermal cells. Our findings offer new mechanistic insights into distinct functions of epidermal versus myeloid cell-derived VEGF-A during HPV-mediated tumorigenesis, with possible implications for preventing this disease.
in vivo blocking of VEGF/VEGFR-2 signaling
Lee, H. J., et al. (2015). "Inhibition of vascular endothelial growth factor A and hypoxia-inducible factor 1alpha maximizes the effects of radiation in sarcoma mouse models through destruction of tumor vasculature" Int J Radiat Oncol Biol Phys 91(3): 621-630. PubMed
PURPOSE: To examine the addition of genetic or pharmacologic inhibition of hypoxia-inducible factor 1alpha (HIF-1alpha) to radiation therapy (RT) and vascular endothelial growth factor A (VEGF-A) inhibition (ie trimodality therapy) for soft-tissue sarcoma. METHODS AND MATERIALS: Hypoxia-inducible factor 1alpha was inhibited using short hairpin RNA or low metronomic doses of doxorubicin, which blocks HIF-1alpha binding to DNA. Trimodality therapy was examined in a mouse xenograft model and a genetically engineered mouse model of sarcoma, as well as in vitro in tumor endothelial cells (ECs) and 4 sarcoma cell lines. RESULTS: In both mouse models, any monotherapy or bimodality therapy resulted in tumor growth beyond 250 mm(3) within the 12-day treatment period, but trimodality therapy with RT, VEGF-A inhibition, and HIF-1alpha inhibition kept tumors at <250 mm(3) for up to 30 days. Trimodality therapy on tumors reduced HIF-1alpha activity as measured by expression of nuclear HIF-1alpha by 87% to 95% compared with RT alone, and cytoplasmic carbonic anhydrase 9 by 79% to 82%. Trimodality therapy also increased EC-specific apoptosis 2- to 4-fold more than RT alone and reduced microvessel density by 75% to 82%. When tumor ECs were treated in vitro with trimodality therapy under hypoxia, there were significant decreases in proliferation and colony formation and increases in DNA damage (as measured by Comet assay and gammaH2AX expression) and apoptosis (as measured by cleaved caspase 3 expression). Trimodality therapy had much less pronounced effects when 4 sarcoma cell lines were examined in these same assays. CONCLUSIONS: Inhibition of HIF-1alpha is highly effective when combined with RT and VEGF-A inhibition in blocking sarcoma growth by maximizing DNA damage and apoptosis in tumor ECs, leading to loss of tumor vasculature.
in vivo blocking of VEGF/VEGFR-2 signaling
Arulanandam, R., et al. (2015). "VEGF-Mediated Induction of PRD1-BF1/Blimp1 Expression Sensitizes Tumor Vasculature to Oncolytic Virus Infection" Cancer Cell 28(2): 210-224. PubMed
Oncolytic viruses designed to attack malignant cells can in addition infect and destroy tumor vascular endothelial cells. We show here that this expanded tropism of oncolytic vaccinia virus to the endothelial compartment is a consequence of VEGF-mediated suppression of the intrinsic antiviral response. VEGF/VEGFR2 signaling through Erk1/2 and Stat3 leads to upregulation, nuclear localization, and activation of the transcription repressor PRD1-BF1/Blimp1. PRD1-BF1 does not contribute to the mitogenic effects of VEGF, but directly represses genes involved in type I interferon (IFN)-mediated antiviral signaling. In vivo suppression of VEGF signaling diminishes PRD1-BF1/Blimp1 expression in tumor vasculature and inhibits intravenously administered oncolytic vaccinia delivery to and consequent spread within the tumor.
in vivo blocking of VEGF/VEGFR-2 signaling, in vitro blocking of VEGFR signaling
Larrayoz, M., et al. (2014). "Contrasting responses of non-small cell lung cancer to antiangiogenic therapies depend on histological subtype" EMBO Mol Med 6(4): 539-550. PubMed
The vascular endothelial growth factor (VEGF) pathway is a clinically validated antiangiogenic target for non-small cell lung cancer (NSCLC). However, some contradictory results have been reported on the biological effects of antiangiogenic drugs. In order to evaluate the efficacy of these drugs in NSCLC histological subtypes, we analyzed the anticancer effect of two anti-VEGFR2 therapies (sunitinib and DC101) in chemically induced mouse models and tumorgrafts of lung adenocarcinoma (ADC) and squamous cell carcinoma (SCC). Antiangiogenic treatments induced vascular trimming in both histological subtypes. In ADC tumors, vascular trimming was accompanied by tumor stabilization. In contrast, in SCC tumors, antiangiogenic therapy was associated with disease progression and induction of tumor proliferation. Moreover, in SCC, anti-VEGFR2 therapies increased the expression of stem cell markers such as aldehyde dehydrogenase 1A1, CD133, and CD15, independently of intratumoral hypoxia. In vitro studies with ADC cell lines revealed that antiangiogenic treatments reduced pAKT and pERK signaling and inhibited proliferation, while in SCC-derived cell lines the same treatments increased pAKT and pERK, and induced survival. In conclusion, this study evaluates for the first time the effect of antiangiogenic drugs in lung SCC murine models in vivo and sheds light on the contradictory results of antiangiogenic therapies in NSCLC.
in vivo blocking of VEGF/VEGFR-2 signaling
Kizhatil, K., et al. (2014). "Schlemm’s canal is a unique vessel with a combination of blood vascular and lymphatic phenotypes that forms by a novel developmental process" PLoS Biol 12(7): e1001912. PubMed
Schlemm’s canal (SC) plays central roles in ocular physiology. These roles depend on the molecular phenotypes of SC endothelial cells (SECs). Both the specific phenotype of SECs and development of SC remain poorly defined. To allow a modern and extensive analysis of SC and its origins, we developed a new whole-mount procedure to visualize its development in the context of surrounding tissues. We then applied genetic lineage tracing, specific-fluorescent reporter genes, immunofluorescence, high-resolution confocal microscopy, and three-dimensional (3D) rendering to study SC. Using these techniques, we show that SECs have a unique phenotype that is a blend of both blood and lymphatic endothelial cell phenotypes. By analyzing whole mounts of postnatal mouse eyes progressively to adulthood, we show that SC develops from blood vessels through a newly discovered process that we name “canalogenesis.” Functional inhibition of KDR (VEGFR2), a critical receptor in initiating angiogenesis, shows that this receptor is required during canalogenesis. Unlike angiogenesis and similar to stages of vasculogenesis, during canalogenesis tip cells divide and form branched chains prior to vessel formation. Differing from both angiogenesis and vasculogenesis, during canalogenesis SECs express Prox1, a master regulator of lymphangiogenesis and lymphatic phenotypes. Thus, SC development resembles a blend of vascular developmental programs. These advances define SC as a unique vessel with a combination of blood vascular and lymphatic phenotypes. They are important for dissecting its functions that are essential for ocular health and normal vision.
in vivo blocking of VEGF/VEGFR-2 signaling
Villalta, S. A., et al. (2013). "Inhibition of VEGFR-2 reverses type 1 diabetes in NOD mice by abrogating insulitis and restoring islet function" Diabetes 62(8): 2870-2878. PubMed
The dysregulation of receptor tyrosine kinases (RTKs) in multiple cell types during chronic inflammation is indicative of their pathogenic role in autoimmune diseases. Among the many RTKs, vascular endothelial growth factor receptor (VEGFR) stands out for its multiple effects on immunity, vascularization, and cell migration. Herein, we examined whether VEGFR participated in the pathogenesis of type 1 diabetes (T1D) in nonobese diabetic (NOD) mice. We found that RTK inhibitors (RTKIs) and VEGF or VEGFR-2 antibodies reversed diabetes when administered at the onset of hyperglycemia. Increased VEGF expression promoted islet vascular remodeling in NOD mice, and inhibition of VEGFR activity with RTKIs abrogated the increase in islet vascularity, impairing T-cell migration into the islet and improving glucose control. Metabolic studies confirmed that RTKIs worked by preserving islet function, as treated mice had improved glucose tolerance without affecting insulin sensitivity. Finally, examination of human pancreata from patients with T1D revealed that VEGFR-2 was confined to the islet vascularity, which was increased in inflamed islets. Collectively, this work reveals a previously unappreciated role for VEGFR-2 signaling in the pathogenesis of T1D by controlling T-cell accessibility to the pancreatic islets and highlights a novel application of VEGFR-2 antagonists for the therapeutic treatment of T1D.
in vivo blocking of VEGF/VEGFR-2 signaling
Chatterjee, S., et al. (2013). "Junctional adhesion molecule-A regulates vascular endothelial growth factor receptor-2 signaling-dependent mouse corneal wound healing" PLoS One 8(5): e63674. PubMed
Inflammation and angiogenesis are integral parts of wound healing. However, excessive and persistent wound-induced inflammation and angiogenesis in an avascular tissue such as the cornea may be associated with scarring and visual impairment. Junctional adhesion molecule A (Jam-A) is a tight junction protein that regulates leukocyte transmigration as well as fibroblast growth factor-2 (FGF-2)-induced angiogenesis. However its function in wound-induced inflammation and angiogenesis is still unknown. In this study, we report spontaneous corneal opacity in Jam-A deficient mice associated with inflammation, angiogenesis and the presence of myofibroblasts. Since wounds and/or corneal infections cause corneal opacities, we tested the role of Jam-A in wound-induced inflammation, angiogenesis and scarring by subjecting Jam-A deficient mice to full thickness corneal wounding. Analysis of these wounds demonstrated increased inflammation, angiogenesis, and increased number of myofibroblasts thereby indicating that Jam-A regulates the wound-healing response by controlling wound-induced inflammation, angiogenesis and scarring in the cornea. These effects were not due to inflammation alone since the inflammation-induced wound-healing response in Jam-A deficient mice was similar to wild type mice. In order to determine the molecular mechanism associated with the observed aberrant corneal wound healing in Jam-A deficient mice, we assessed the expression of the components of vascular endothelial growth factor A (VEGF-A)/vascular endothelial growth factor receptor- 2(VEGFR-2) signaling pathway. Interestingly, we observed increased levels of VEGF-A mRNA in Jam-A deficient eyes. We also observed nuclear localization of phosphorylated SMAD3 (pSMAD3) indicative of TGFbeta pathway activation in the Jam-A deficient eyes. Furthermore the increased wound-induced corneal inflammation, angiogenesis, and scarring in Jam-A deficient mice was attenuated by treatment with DC101, an anti-vascular endothelial growth factor receptor-2 (VEGFR-2) antibody. Our results suggest that in the absence of Jam-A, the VEGF-A/VEGFR-2 pathway is upregulated, thereby augmenting wound induced corneal inflammation, angiogenesis, and myofibroblast accumulation leading to scarring.
in vivo blocking of VEGF/VEGFR-2 signaling
Kim, Y. J., et al. (2013). "Overcoming evasive resistance from vascular endothelial growth factor a inhibition in sarcomas by genetic or pharmacologic targeting of hypoxia-inducible factor 1alpha" Int J Cancer 132(1): 29-41. PubMed
Increased levels of hypoxia and hypoxia-inducible factor 1alpha (HIF-1alpha) in human sarcomas correlate with tumor progression and radiation resistance. Prolonged antiangiogenic therapy of tumors not only delays tumor growth but may also increase hypoxia and HIF-1alpha activity. In our recent clinical trial, treatment with the vascular endothelial growth factor A (VEGF-A) antibody, bevacizumab, followed by a combination of bevacizumab and radiation led to near complete necrosis in nearly half of sarcomas. Gene Set Enrichment Analysis of microarrays from pretreatment biopsies found that the Gene Ontology category “Response to hypoxia” was upregulated in poor responders and that the hierarchical clustering based on 140 hypoxia-responsive genes reliably separated poor responders from good responders. The most commonly used chemotherapeutic drug for sarcomas, doxorubicin (Dox), was recently found to block HIF-1alpha binding to DNA at low metronomic doses. In four sarcoma cell lines, HIF-1alpha shRNA or Dox at low concentrations blocked HIF-1alpha induction of VEGF-A by 84-97% and carbonic anhydrase 9 by 83-93%. HT1080 sarcoma xenografts had increased hypoxia and/or HIF-1alpha activity with increasing tumor size and with anti-VEGF receptor antibody (DC101) treatment. Combining DC101 with HIF-1alpha shRNA or metronomic Dox had a synergistic effect in suppressing growth of HT1080 xenografts, at least in part via induction of tumor endothelial cell apoptosis. In conclusion, sarcomas respond to increased hypoxia by expressing HIF-1alpha target genes that may promote resistance to antiangiogenic and other therapies. HIF-1alpha inhibition blocks this evasive resistance and augments destruction of the tumor vasculature.
in vivo blocking of VEGF/VEGFR-2 signaling
Kumar, V., et al. (2010). "Global lymphoid tissue remodeling during a viral infection is orchestrated by a B cell-lymphotoxin-dependent pathway" Blood 115(23): 4725-4733. PubMed
Adaptive immune responses are characterized by substantial restructuring of secondary lymphoid organs. The molecular and cellular factors responsible for virus-induced lymphoid remodeling are not well known to date. Here we applied optical projection tomography, a mesoscopic imaging technique, for a global analysis of the entire 3-dimensional structure of mouse peripheral lymph nodes (PLNs), focusing on B-cell areas and high endothelial venule (HEV) networks. Structural homeostasis of PLNs was characterized by a strict correlation between total PLN volume, B-cell volume, B-cell follicle number, and HEV length. After infection with lymphocytic choriomeningitis virus, we observed a substantial, lymphotoxin (LT) beta-receptor-dependent reorganization of the PLN microarchitecture, in which an initial B-cell influx was followed by 3-fold increases in PLN volume and HEV network length on day 8 after infection. Adoptive transfer experiments revealed that virus-induced PLN and HEV network remodeling required LTalpha(1)beta(2)-expressing B cells, whereas the inhibition of vascular endothelial growth factor-A signaling pathways had no significant effect on PLN expansion. In summary, lymphocytic choriomeningitis virus-induced PLN growth depends on a vascular endothelial growth factor-A-independent, LT- and B cell-dependent morphogenic pathway, as revealed by an in-depth mesoscopic analysis of the global PLN structure.
in vivo blocking of VEGF/VEGFR-2 signaling
Kilarski, W. W., et al. (2009). "Biomechanical regulation of blood vessel growth during tissue vascularization" Nat Med 15(6): 657-664. PubMed
Formation of new vessels in granulation tissue during wound healing has been assumed to occur solely through sprouting angiogenesis. In contrast, we show here that neovascularization can be accomplished by nonangiogenic expansion of preexisting vessels. Using neovascularization models based on the chick chorioallantoic membrane and the healing mouse cornea, we found that tissue tension generated by activated fibroblasts or myofibroblasts during wound contraction mediated and directed translocation of the vasculature. These mechanical forces pulled vessels from the preexisting vascular bed as vascular loops with functional circulation that expanded as an integral part of the growing granulation tissue through vessel enlargement and elongation. Blockade of vascular endothelial growth factor receptor-2 confirmed that biomechanical forces were sufficient to mediate the initial vascular growth independently of endothelial sprouting or proliferation. The neovascular network was further remodeled by splitting, sprouting and regression of individual vessels. This model explains the rapid appearance of large functional vessels in granulation tissue during wound healing.