InVivoMAb anti-mouse CD8β (Lyt 3.2)
Product Details
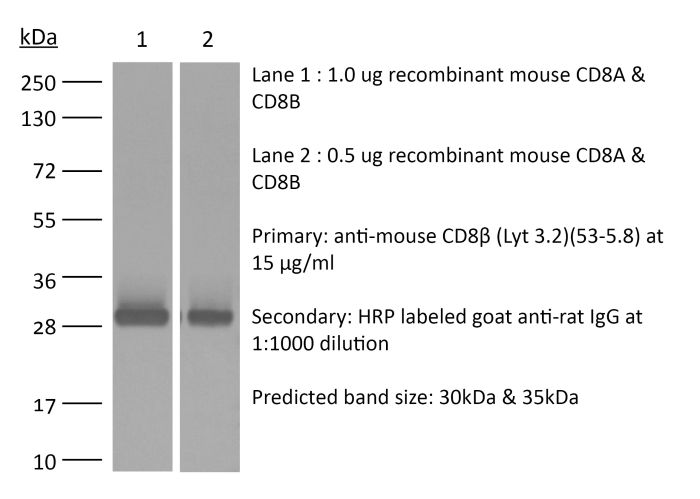
The 53-5.8 monoclonal antibody reacts with mouse CD8β also known as Lyt 3.2. The CD8 antigen is a transmembrane glycoprotein that acts as a co-receptor for the T cell receptor (TCR). Like the TCR, CD8 binds to class I MHC molecules displayed by antigen presenting cells (APC). CD8 is primarily expressed on the surface of cytotoxic T cells, but can also be found on thymocytes, natural killer cells, and some dendritic cell subsets. CD8 most commonly exists as a heterodimer composed of one CD8α and one CD8β chain however, it can also exist as a homodimer composed of two CD8α chains. Both the CD8α and CD8β chains share significant homology to immunoglobulin variable light chains. The molecular weight of each CD8 chain is approximately 34 kDa. The 53-5.8 antibody has been shown to deplete CD8+ T cells completely but not deplete CD8+ CD11c+ dendritic cells when used in vivo.Specifications
| Isotype | Rat IgG1, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb rat IgG1 isotype control, anti-horseradish peroxidase |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Mouse thymus or spleen |
| Reported Applications |
in vivo CD8+ T cell depletion in vitro CD8 blockade Immunofluorescence |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
<2EU/mg (<0.002EU/μg) Determined by LAL gel clotting assay |
| Purity |
>95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_2687706 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
Recommended Products
-

Recommended Isotype Control(s)
InVivoMAb rat IgG1 isotype control, anti-horseradish peroxidase
-

Recommended Dilution Buffer
InVivoPure pH 7.0 Dilution Buffer
Immunofluorescence
Howland, S. W., et al. (2015). "Activated Brain Endothelial Cells Cross-Present Malaria Antigen" PLoS Pathog 11(6): e1004963. PubMed
In the murine model of cerebral malaria caused by P. berghei ANKA (PbA), parasite-specific CD8+ T cells directly induce pathology and have long been hypothesized to kill brain endothelial cells that have internalized PbA antigen. We previously reported that brain microvessel fragments from infected mice cross-present PbA epitopes, using reporter cells transduced with epitope-specific T cell receptors. Here, we confirm that endothelial cells are the population responsible for cross-presentation in vivo, not pericytes or microglia. PbA antigen cross-presentation by primary brain endothelial cells in vitro confers susceptibility to killing by CD8+ T cells from infected mice. IFNgamma stimulation is required for brain endothelial cross-presentation in vivo and in vitro, which occurs by a proteasome- and TAP-dependent mechanism. Parasite strains that do not induce cerebral malaria were phagocytosed and cross-presented less efficiently than PbA in vitro. The main source of antigen appears to be free merozoites, which were avidly phagocytosed. A human brain endothelial cell line also phagocytosed P. falciparum merozoites. Besides being the first demonstration of cross-presentation by brain endothelial cells, our results suggest that interfering with merozoite phagocytosis or antigen processing may be effective strategies for cerebral malaria intervention.
in vivo CD8+ T cell depletion
Guillerey, C., et al. (2015). "Immunosurveillance and therapy of multiple myeloma are CD226 dependent" J Clin Invest 125(5): 2077-2089. PubMed
Multiple myeloma (MM) is an age-dependent hematological malignancy. Evaluation of immune interactions that drive MM relies on in vitro experiments that do not reflect the complex cellular stroma involved in MM pathogenesis. Here we used Vk*MYC transgenic mice, which spontaneously develop MM, and demonstrated that the immune system plays a critical role in the control of MM progression and the response to treatment. We monitored Vk*MYC mice that had been crossed with Cd226 mutant mice over a period of 3 years and found that CD226 limits spontaneous MM development. The CD226-dependent anti-myeloma immune response against transplanted Vk*MYC MM cells was mediated both by NK and CD8+ T cells through perforin and IFN-gamma pathways. Moreover, CD226 expression was required for optimal antimyeloma efficacy of cyclophosphamide (CTX) and bortezomib (Btz), which are both standardly used to manage MM in patients. Activation of costimulatory receptor CD137 with mAb (4-1BB) exerted strong antimyeloma activity, while inhibition of coinhibitory receptors PD-1 and CTLA-4 had no effect. Taken together, the results of this study provide in vivo evidence that CD226 is important for MM immunosurveillance and indicate that specific immune components should be targeted for optimal MM treatment efficacy. As progressive immunosuppression associates with MM development, strategies aimed to increase immune functions may have important therapeutic implications in MM.
in vivo CD8+ T cell depletion
Kobayashi, T., et al. (2015). "NKT cell-targeted vaccination plus anti-4-1BB antibody generates persistent CD8 T cell immunity against B cell lymphoma" Oncoimmunology 4(3): e990793. PubMed
Harnessing the immune adjuvant properties of natural killer T (NKT) cells is an effective strategy to generate anticancer immunity. The objective of this study was to increase the potency and durability of vaccine-induced immunity against B cell lymphoma by combining alpha-galactosylceramide (alpha-GalCer)-loaded tumor cell vaccination with an agonistic antibody targeting the immune checkpoint molecule 4-1BB (CD137). We observed potent synergy when combining vaccination and anti-4-1BB antibody treatment resulting in significantly enhanced survival of mice harboring Emu-myc tumors, including complete eradication of lymphoma in over 50% of mice. Tumor-free survival required interferon gamma (IFNgamma)-dependent expansion of CD8+ T cells and was associated with 4-1BB-mediated differentiation of KLRG1+ effector CD8+ T cells. ‘Cured’ mice were also resistant to lymphoma re-challenge 80 days later indicating successful generation of immunological memory. Overall, our results demonstrate that therapeutic anticancer vaccination against B cell lymphoma using an NKT cell ligand can be boosted by subsequent co-stimulation through 4-1BB leading to a sustainable immune response that may enhance outcomes to conventional treatment.
in vivo CD8+ T cell depletion
Allard, B., et al. (2013). "Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs" Clin Cancer Res 19(20): 5626-5635. PubMed
PURPOSE: Monoclonal antibodies (mAb) that block programmed death (PD)-1 or cytotoxic T lymphocyte antigen (CTLA-4) receptors have been associated with durable clinical responses against a variety of cancer types and hold great potential as novel cancer therapeutics. Recent evidence suggest that targeted blockade of multiple immunosuppressive pathways can induce synergistic antitumor responses. EXPERIMENTAL DESIGN: In this study, we investigated whether targeted blockade of CD73, an ectonucleotidase that catabolizes the hydrolysis of extracellular adenosine monophosphate (AMP) to adenosine, can enhance the antitumor activity of anti-CTLA-4 and anti-PD-1 mAbs against transplanted and chemically induced mouse tumors. RESULTS: Anti-CD73 mAb significantly enhanced the activity of both anti-CTLA-4 and anti-PD-1 mAbs against MC38-OVA (colon) and RM-1 (prostate) subcutaneous tumors, and established metastatic 4T1.2 breast cancer. Anti-CD73 mAb also significantly enhanced the activity of anti-PD-1 mAb against 3-methylcholanthrene (MCA)-induced fibrosarcomas. Gene-targeted mice revealed that single-agent therapies and combinatorial treatments were dependent on host IFN-gamma and CD8(+) T cells, but independent of perforin. Interestingly, anti-CD73 mAb preferentially synergized with anti-PD-1 mAb. We investigated the effect of extracellular adenosine on tumor-infiltrating T cells and showed that activation of A2A adenosine receptor enhances PD-1 expression, but not CTLA-4 expression, on tumor-specific CD8+ T cells and CD4+ Foxp3+ T regulatory cells. CONCLUSIONS: Taken together, our study revealed that targeted blockade of CD73 can enhance the therapeutic activity of anti-PD-1 and anti-CTLA-4 mAbs and may thus potentiate therapeutic strategies targeting immune checkpoint inhibitors in general.
in vivo CD8+ T cell depletion
Ahmed, K. A., et al. (2012). "Direct in vivo evidence of CD4+ T cell requirement for CTL response and memory via pMHC-I targeting and CD40L signaling" J Leukoc Biol 92(2): 289-300. PubMed
CD4(+) T cell help contributes critically to DC-induced CD8(+) CTL immunity. However, precisely how these three cell populations interact and how CD4(+) T cell signals are delivered to CD8(+) T cells in vivo have been unclear. In this study, we developed a novel, two-step approach, wherein CD4(+) T cells and antigen-presenting DCs productively engaged one another in vivo in the absence of cognate CD8(+) T cells, after which, we selectively depleted the previously engaged CD4(+) T cells or DCs before allowing interactions of either population alone with naive CD8(+) T cells. This protocol thus allows us to clearly document the importance of CD4(+) T-licensed DCs and DC-primed CD4(+) T cells in CTL immunity. Here, we provide direct in vivo evidence that primed CD4(+) T cells or licensed DCs can stimulate CTL response and memory, independent of DC-CD4(+) T cell clusters. Our results suggest that primed CD4(+) T cells with acquired pMHC-I from DCs represent crucial “immune intermediates” for rapid induction of CTL responses and for functional memory via CD40L signaling. Importantly, intravital, two-photon microscopy elegantly provide unequivocal in vivo evidence for direct CD4-CD8(+) T cell interactions via pMHC-I engagement. This study corroborates the coexistence of direct and indirect mechanisms of T cell help for a CTL response in noninflammatory situations. These data suggest a new “dynamic model of three-cell interactions” for CTL immunity derived from stimulation by dissociated, licensed DCs, primed CD4(+) T cells, and DC-CD4(+) T cell clusters and may have significant implications for autoimmunity and vaccine design.
in vivo CD8+ T cell depletion
Verbrugge, I., et al. (2012). "Radiotherapy increases the permissiveness of established mammary tumors to rejection by immunomodulatory antibodies" Cancer Res 72(13): 3163-3174. PubMed
It is becoming increasingly evident that radiotherapy may benefit from coincident or subsequent immunotherapy. In this study, we examined whether the antitumor effects of radiotherapy, in established triple-negative breast tumors could be enhanced with combinations of clinically relevant monoclonal antibodies (mAb), designed to stimulate immunity [anti-(alpha)-CD137, alpha-CD40] or relieve immunosuppression [alpha-programmed death (PD)-1]. While the concomitant targeting of the costimulatory molecules CD137 and CD40 enhanced the antitumor effects of radiotherapy and promoted the rejection of subcutaneous BALB/c-derived 4T1.2 tumors, this novel combination was noncurative in mice bearing established C57BL/6-derived AT-3 tumors. We identified PD-1 signaling within the AT-3 tumors as a critical limiting factor to the therapeutic efficacy of alpha-CD137 therapy, alone and in combination with radiotherapy. Strikingly, all mice bearing established orthotopic AT-3 mammary tumors were cured when alpha-CD137 and alpha-PD-1 mAbs were combined with single- or low-dose fractionated radiotherapy. CD8+ T cells were essential for curative responses to this combinatorial regime. Interestingly, CD137 expression on tumor-associated CD8+ T cells was largely restricted to a subset that highly expressed PD-1. These CD137+PD-1High CD8+ T cells, persisted in irradiated AT-3 tumors, expressed Tim-3, granzyme B and Ki67 and produced IFN-gamma ex vivo in response to phorbol 12-myristate 13-acetate (PMA) and ionomycin stimulation. Notably, radiotherapy did not deplete, but enriched tumors of functionally active, tumor-specific effector cells. Collectively, these data show that concomitant targeting of immunostimulatory and inhibitory checkpoints with immunomodulatory mAbs can enhance the curative capacity of radiotherapy in established breast malignancy.
in vitro CD8 blockade
Takada, K. and S. C. Jameson. (2009). "Self-class I MHC molecules support survival of naive CD8 T cells, but depress their functional sensitivity through regulation of CD8 expression levels" J Exp Med 206(10): 2253-2269. PubMed
Previous studies have suggested that naive CD8 T cells require self-peptide-major histocompatability complex (MHC) complexes for maintenance. However, interpretation of such studies is complicated because of the involvement of lymphopenic animals, as lymphopenia drastically alters naive T cell homeostasis and function. In this study, we explored naive CD8 T cell survival and function in nonlymphopenic conditions by using bone marrow chimeric donors and hosts in which class I MHC expression is absent or limited to radiosensitive versus radioresistant cells. We found that long-term survival of naive CD8 T cells (but not CD4 T cells) was impaired in the absence of class I MHC. However, distinct from this effect, class I MHC deprivation also enhanced naive CD8 T cell responsiveness to low-affinity (but not high-affinity) peptide-MHC ligands. We found that this improved sensitivity was a consequence of up-regulated CD8 levels, which was mediated through a transcriptional mechanism. Hence, our data suggest that, in a nonlymphopenic setting, self-class I MHC molecules support CD8 T cell survival, but that these interactions also attenuate naive T cell sensitivity by dynamic tuning of CD8 levels.