InVivoMAb anti-mouse CD8α

Product Details
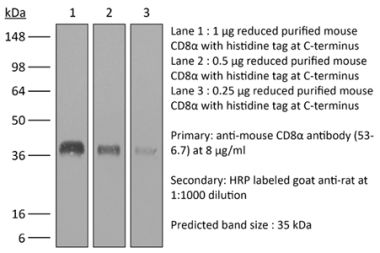
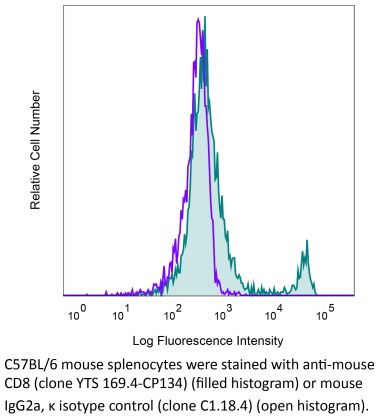
The YTS 169.4 monoclonal antibody reacts with mouse CD8α. The CD8 antigen is a transmembrane glycoprotein that acts as a co-receptor for the T cell receptor (TCR). Like the TCR, CD8 binds to class I MHC molecules displayed by antigen presenting cells (APC). CD8 is primarily expressed on the surface of cytotoxic T cells, but can also be found on thymocytes, natural killer cells, and some dendritic cell subsets. CD8 most commonly exists as a heterodimer composed of one CD8α and one CD8β chain however, it can also exist as a homodimer composed of two CD8α chains. Both the CD8α and CD8β chains share significant homology to immunoglobulin variable light chains. The molecular weight of each CD8 chain is approximately 34 kDa. The YTS 169.4 antibody exhibits depleting activity when used in vivo.Specifications
| Isotype | Rat IgG2b, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb rat IgG2b isotype control, anti-keyhole limpet hemocyanin |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | CBA mouse thymocytes |
| Reported Applications |
in vivo CD8+ T cell depletion Western blot |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
<2EU/mg (<0.002EU/μg) Determined by LAL gel clotting assay |
| Purity |
>95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_10950145 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
Additional Formats


Recommended Products
-

Recommended Isotype Control(s)
InVivoMAb rat IgG2b isotype control, anti-keyhole limpet hemocyanin
-

Recommended Dilution Buffer
InVivoPure pH 7.0 Dilution Buffer
in vivo CD8+ T cell depletion
Vashist, N., et al. (2018). "Influenza-Activated ILC1s Contribute to Antiviral Immunity Partially Influenced by Differential GITR Expression" Front Immunol 9: 505. PubMed
Innate lymphoid cells (ILCs) represent diversified subsets of effector cells as well as immune regulators of mucosal immunity and are classified into group 1 ILCs, group 2 ILCs, and group 3 ILCs. Group 1 ILCs encompass natural killer (NK) cells and non-NK ILCs (ILC1s) and mediate their functionality via the rapid production of IFN-gamma and TNF-alpha. The current knowledge of ILC1s mainly associates them to inflammatory processes. Much less is known about their regulation during infection and their capacity to interact with cells of the adaptive immune system. The present study dissected the role of ILC1s during early influenza A virus infection, thereby revealing their impact on the antiviral response. Exploiting in vitro and in vivo H1N1 infection systems, a cross-talk of ILC1s with cells of the innate and the adaptive immunity was demonstrated, which contributes to anti-influenza immunity. A novel association of ILC1 functionality and the expression of the glucocorticoid-induced TNFR-related protein (GITR) was observed, which hints toward a so far undescribed role of GITR in regulating ILC1 responsiveness. Overexpression of GITR inhibits IFN-gamma production by ILC1s, whereas partial reduction of GITR expression can reverse this effect, thereby regulating ILC1 functionality. These new insights into ILC1 biology define potential intervention targets to modulate the functional properties of ILC1s, thus contributing toward the development of new immune interventions against influenza.
in vivo CD8+ T cell depletion
Triplett, T. A., et al. (2018). "Reversal of indoleamine 2,3-dioxygenase-mediated cancer immune suppression by systemic kynurenine depletion with a therapeutic enzyme" Nat Biotechnol 36(8): 758-764. PubMed
Increased tryptophan (Trp) catabolism in the tumor microenvironment (TME) can mediate immune suppression by upregulation of interferon (IFN)-gamma-inducible indoleamine 2,3-dioxygenase (IDO1) and/or ectopic expression of the predominantly liver-restricted enzyme tryptophan 2,3-dioxygenase (TDO). Whether these effects are due to Trp depletion in the TME or mediated by the accumulation of the IDO1 and/or TDO (hereafter referred to as IDO1/TDO) product kynurenine (Kyn) remains controversial. Here we show that administration of a pharmacologically optimized enzyme (PEGylated kynureninase; hereafter referred to as PEG-KYNase) that degrades Kyn into immunologically inert, nontoxic and readily cleared metabolites inhibits tumor growth. Enzyme treatment was associated with a marked increase in the tumor infiltration and proliferation of polyfunctional CD8(+) lymphocytes. We show that PEG-KYNase administration had substantial therapeutic effects when combined with approved checkpoint inhibitors or with a cancer vaccine for the treatment of large B16-F10 melanoma, 4T1 breast carcinoma or CT26 colon carcinoma tumors. PEG-KYNase mediated prolonged depletion of Kyn in the TME and reversed the modulatory effects of IDO1/TDO upregulation in the TME.
in vivo CD8+ T cell depletion
Carmi, Y., et al. (2015). "Allogeneic IgG combined with dendritic cell stimuli induce antitumour T-cell immunity" Nature 521(7550): 99-104. PubMed
Whereas cancers grow within host tissues and evade host immunity through immune-editing and immunosuppression, tumours are rarely transmissible between individuals. Much like transplanted allogeneic organs, allogeneic tumours are reliably rejected by host T cells, even when the tumour and host share the same major histocompatibility complex alleles, the most potent determinants of transplant rejection. How such tumour-eradicating immunity is initiated remains unknown, although elucidating this process could provide the basis for inducing similar responses against naturally arising tumours. Here we find that allogeneic tumour rejection is initiated in mice by naturally occurring tumour-binding IgG antibodies, which enable dendritic cells (DCs) to internalize tumour antigens and subsequently activate tumour-reactive T cells. We exploited this mechanism to treat autologous and autochthonous tumours successfully. Either systemic administration of DCs loaded with allogeneic-IgG-coated tumour cells or intratumoral injection of allogeneic IgG in combination with DC stimuli induced potent T-cell-mediated antitumour immune responses, resulting in tumour eradication in mouse models of melanoma, pancreas, lung and breast cancer. Moreover, this strategy led to eradication of distant tumours and metastases, as well as the injected primary tumours. To assess the clinical relevance of these findings, we studied antibodies and cells from patients with lung cancer. T cells from these patients responded vigorously to autologous tumour antigens after culture with allogeneic-IgG-loaded DCs, recapitulating our findings in mice. These results reveal that tumour-binding allogeneic IgG can induce powerful antitumour immunity that can be exploited for cancer immunotherapy.
in vivo CD8+ T cell depletion
Burrack, K. S., et al. (2015). "Myeloid Cell Arg1 Inhibits Control of Arthritogenic Alphavirus Infection by Suppressing Antiviral T Cells" PLoS Pathog 11(10): e1005191. PubMed
Arthritogenic alphaviruses, including Ross River virus (RRV) and chikungunya virus (CHIKV), are responsible for explosive epidemics involving millions of cases. These mosquito-transmitted viruses cause inflammation and injury in skeletal muscle and joint tissues that results in debilitating pain. We previously showed that arginase 1 (Arg1) was highly expressed in myeloid cells in the infected and inflamed musculoskeletal tissues of RRV- and CHIKV-infected mice, and specific deletion of Arg1 from myeloid cells resulted in enhanced viral control. Here, we show that Arg1, along with other genes associated with suppressive myeloid cells, is induced in PBMCs isolated from CHIKV-infected patients during the acute phase as well as the chronic phase, and that high Arg1 expression levels were associated with high viral loads and disease severity. Depletion of both CD4 and CD8 T cells from RRV-infected Arg1-deficient mice restored viral loads to levels detected in T cell-depleted wild-type mice. Moreover, Arg1-expressing myeloid cells inhibited virus-specific T cells in the inflamed and infected musculoskeletal tissues, but not lymphoid tissues, following RRV infection in mice, including suppression of interferon-gamma and CD69 expression. Collectively, these data enhance our understanding of the immune response following arthritogenic alphavirus infection and suggest that immunosuppressive myeloid cells may contribute to the duration or severity of these debilitating infections.
in vivo CD8+ T cell depletion
Wensveen, F. M., et al. (2015). "NK cells link obesity-induced adipose stress to inflammation and insulin resistance" Nat Immunol 16(4): 376-385. PubMed
An important cause of obesity-induced insulin resistance is chronic systemic inflammation originating in visceral adipose tissue (VAT). VAT inflammation is associated with the accumulation of proinflammatory macrophages in adipose tissue, but the immunological signals that trigger their accumulation remain unknown. We found that a phenotypically distinct population of tissue-resident natural killer (NK) cells represented a crucial link between obesity-induced adipose stress and VAT inflammation. Obesity drove the upregulation of ligands of the NK cell-activating receptor NCR1 on adipocytes; this stimulated NK cell proliferation and interferon-gamma (IFN-gamma) production, which in turn triggered the differentiation of proinflammatory macrophages and promoted insulin resistance. Deficiency of NK cells, NCR1 or IFN-gamma prevented the accumulation of proinflammatory macrophages in VAT and greatly ameliorated insulin sensitivity. Thus NK cells are key regulators of macrophage polarization and insulin resistance in response to obesity-induced adipocyte stress.
in vivo CD8+ T cell depletion
Li, Z., et al. (2015). "Pre-treatment of allogeneic bone marrow recipients with the CXCR4 antagonist AMD3100 transiently enhances hematopoietic chimerism without promoting donor-specific skin allograft tolerance" Transpl Immunol 33(2): 125-129. PubMed
Hematopoietic chimerism established by allogeneic bone marrow transplantation is known to promote donor-specific organ allograft tolerance; however, clinical application is limited by the need for toxic host conditioning and “megadoses” of donor bone marrow cells. A potential solution to this problem has been suggested by the observation that recipient bone marrow mobilization by the CXCR4 antagonist AMD3100 promotes chimerism in congenic bone marrow transplantation experiments in mice. Here we report that a single subcutaneous dose of 10mg/kg AMD3100 in recipient C57BL/6 mice was able to enhance hematopoietic chimerism when complete MHC-mismatched BALB/c donor bone marrow cells were transplanted 1h after drug dosing. However, levels of chimerism measured 30days post-transplantation were not sustained when mice were reexamined on day 90 post-transplantation. Moreover, transient chimerism induced by this protocol did not support robust donor-specific skin allograft tolerance. Using the same transient immunosuppression protocol, we confirmed that “megadoses” of donor bone marrow cells could induce durable chimerism associated with donor-specific skin allograft tolerance without AMD3100 pre-treatment. We conclude that in this protocol AMD3100 pretreatment may empty bone marrow niches that become reoccupied by allogeneic donor hematopoietic progenitor cells but not by true long-lived donor hematopoietic stem cells, resulting in short-lived chimerism and failure to support durable donor-specific allograft tolerance.
in vivo CD8+ T cell depletion
Krupnick, A. S., et al. (2014). "Central memory CD8+ T lymphocytes mediate lung allograft acceptance" J Clin Invest 124(3): 1130-1143. PubMed
Memory T lymphocytes are commonly viewed as a major barrier for long-term survival of organ allografts and are thought to accelerate rejection responses due to their rapid infiltration into allografts, low threshold for activation, and ability to produce inflammatory mediators. Because memory T cells are usually associated with rejection, preclinical protocols have been developed to target this population in transplant recipients. Here, using a murine model, we found that costimulatory blockade-mediated lung allograft acceptance depended on the rapid infiltration of the graft by central memory CD8+ T cells (CD44(hi)CD62L(hi)CCR7+). Chemokine receptor signaling and alloantigen recognition were required for trafficking of these memory T cells to lung allografts. Intravital 2-photon imaging revealed that CCR7 expression on CD8+ T cells was critical for formation of stable synapses with antigen-presenting cells, resulting in IFN-gamma production, which induced NO and downregulated alloimmune responses. Thus, we describe a critical role for CD8+ central memory T cells in lung allograft acceptance and highlight the need for tailored approaches for tolerance induction in the lung.
in vivo CD8+ T cell depletion
Pastille, E., et al. (2014). "Transient ablation of regulatory T cells improves antitumor immunity in colitis-associated colon cancer" Cancer Res 74(16): 4258-4269. PubMed
Regulatory T cells (Treg) are supportive to cancer development in most tissues, but their role in colitis-associated colon cancer (CAC) remains unclear. In this study, we investigated the role of CD4(+)Foxp3(+) Treg in a mouse model of CAC and in patients with colon cancer. These Treg were increased strongly in number in a mouse model of CAC and in the peripheral blood of patients with colon cancer, exhibiting an activated phenotype as defined by elevated expression of GARP, CD103, CTLA-4, and IL10, along with an increased suppressive effect on the proliferation and Th1 cytokine expression of CD4(+)CD25(-) responder T cells ex vivo. Transient ablation of CD4(+)Foxp3(+) Treg during tumor development in the CAC model suppressed tumor outgrowth and distribution, accompanied by an increased number of CD8(+)IFNgamma/granzyme B-producing effector T cells. Conversely, inactivation of IL10 in Treg did not elevate the antitumor response but instead further boosted tumor development. Our results establish a tumor-promoting function for Treg during CAC formation, but they also suggest that a selective, transient ablation of Treg can evoke antitumor responses, with implications for immunotherapeutic interventions in patients with CAC.
in vivo CD8+ T cell depletion
Bivas-Benita, M., et al. (2013). "Airway CD8(+) T cells induced by pulmonary DNA immunization mediate protective anti-viral immunity" Mucosal Immunol 6(1): 156-166. PubMed
Vaccination strategies for protection against a number of respiratory pathogens must induce T-cell populations in both the pulmonary airways and peripheral lymphoid organs. In this study, we show that pulmonary immunization using plasmid DNA formulated with the polymer polyethyleneimine (PEI-DNA) induced antigen-specific CD8(+) T cells in the airways that persisted long after antigen local clearance. The persistence of the cells was not mediated by local lymphocyte proliferation or persistent antigen presentation within the lung or airways. These vaccine-induced CD8(+) T cells effectively mediated protective immunity against respiratory challenges with vaccinia virus and influenza virus. Moreover, this protection was not dependent upon the recruitment of T cells from peripheral sites. These findings demonstrate that pulmonary immunization with PEI-DNA is an efficient approach for inducing robust pulmonary CD8(+) T-cell populations that are effective at protecting against respiratory pathogens.
in vivo CD8+ T cell depletion
Dai, M., et al. (2013). "Long-lasting complete regression of established mouse tumors by counteracting Th2 inflammation" J Immunother 36(4): 248-257. PubMed
40% of mice with SW1 tumors remained healthy >150 days after last treatment and are probably cured. Therapeutic efficacy was associated with a systemic immune response with memory and antigen specificity, required CD4 cells and involved CD8 cells and NK cells to a less extent. The 3 mAb combination significantly decreased CD19 cells at tumor sites, increased IFN-gamma and TNF-alpha producing CD4 and CD8 T cells and mature CD86 dendritic cells (DC), and it increased the ratios of effector CD4 and CD8 T cells to CD4Foxp3 regulatory T (Treg) cells and to CD11bGr-1 myeloid suppressor cells (MDSC). This is consistent with shifting the tumor microenvironment from an immunosuppressive Th2 to an immunostimulatory Th1 type and is further supported by PCR data. Adding an anti-CD19 mAb to the 3 mAb combination in the SW1 model further increased therapeutic efficacy. Data from ongoing experiments show that intratumoral injection of a combination of mAbs to CD137PD-1CTLA4CD19 can induce complete regression and dramatically prolong survival also in the TC1 carcinoma and B16 melanoma models, suggesting that the approach has general validity.”}” data-sheets-userformat=”{“2″:14851,”3”:{“1″:0},”4”:{“1″:2,”2″:16777215},”12″:0,”14”:{“1″:2,”2″:1521491},”15″:”Roboto, sans-serif”,”16″:12}”>Mice with intraperitoneal ID8 ovarian carcinoma or subcutaneous SW1 melanoma were injected with monoclonal antibodies (mAbs) to CD137PD-1CTLA4 7-15 days after tumor initiation. Survival of mice with ID8 tumors tripled and >40% of mice with SW1 tumors remained healthy >150 days after last treatment and are probably cured. Therapeutic efficacy was associated with a systemic immune response with memory and antigen specificity, required CD4 cells and involved CD8 cells and NK cells to a less extent. The 3 mAb combination significantly decreased CD19 cells at tumor sites, increased IFN-gamma and TNF-alpha producing CD4 and CD8 T cells and mature CD86 dendritic cells (DC), and it increased the ratios of effector CD4 and CD8 T cells to CD4Foxp3 regulatory T (Treg) cells and to CD11bGr-1 myeloid suppressor cells (MDSC). This is consistent with shifting the tumor microenvironment from an immunosuppressive Th2 to an immunostimulatory Th1 type and is further supported by PCR data. Adding an anti-CD19 mAb to the 3 mAb combination in the SW1 model further increased therapeutic efficacy. Data from ongoing experiments show that intratumoral injection of a combination of mAbs to CD137PD-1CTLA4CD19 can induce complete regression and dramatically prolong survival also in the TC1 carcinoma and B16 melanoma models, suggesting that the approach has general validity.
in vivo CD8+ T cell depletion
Sledzinska, A., et al. (2013). "TGF-beta signalling is required for CD4(+) T cell homeostasis but dispensable for regulatory T cell function" PLoS Biol 11(10): e1001674. PubMed
TGF-beta is widely held to be critical for the maintenance and function of regulatory T (T(reg)) cells and thus peripheral tolerance. This is highlighted by constitutive ablation of TGF-beta receptor (TR) during thymic development in mice, which leads to a lethal autoimmune syndrome. Here we describe that TGF-beta-driven peripheral tolerance is not regulated by TGF-beta signalling on mature CD4(+) T cells. Inducible TR2 ablation specifically on CD4(+) T cells did not result in a lethal autoinflammation. Transfer of these TR2-deficient CD4(+) T cells to lymphopenic recipients resulted in colitis, but not overt autoimmunity. In contrast, thymic ablation of TR2 in combination with lymphopenia led to lethal multi-organ inflammation. Interestingly, deletion of TR2 on mature CD4(+) T cells does not result in the collapse of the T(reg) cell population as observed in constitutive models. Instead, a pronounced enlargement of both regulatory and effector memory T cell pools was observed. This expansion is cell-intrinsic and seems to be caused by increased T cell receptor sensitivity independently of common gamma chain-dependent cytokine signals. The expression of Foxp3 and other regulatory T cells markers was not dependent on TGF-beta signalling and the TR2-deficient T(reg) cells retained their suppressive function both in vitro and in vivo. In summary, absence of TGF-beta signalling on mature CD4(+) T cells is not responsible for breakdown of peripheral tolerance, but rather controls homeostasis of mature T cells in adult mice.
in vivo CD8+ T cell depletion
Krieg, C., et al. (2010). "Improved IL-2 immunotherapy by selective stimulation of IL-2 receptors on lymphocytes and endothelial cells" Proc Natl Acad Sci U S A 107(26): 11906-11911. PubMed
IL-2 immunotherapy is an attractive treatment option for certain metastatic cancers. However, administration of IL-2 to patients can lead, by ill-defined mechanisms, to toxic adverse effects including severe pulmonary edema. Here, we show that IL-2-induced pulmonary edema is caused by direct interaction of IL-2 with functional IL-2 receptors (IL-2R) on lung endothelial cells in vivo. Treatment of mice with high-dose IL-2 led to efficient expansion of effector immune cells expressing high levels of IL-2Rbetagamma, including CD8(+) T cells and natural killer cells, which resulted in a considerable antitumor response against s.c. and pulmonary B16 melanoma nodules. However, high-dose IL-2 treatment also affected immune cell lineage marker-negative CD31(+) pulmonary endothelial cells via binding to functional alphabetagamma IL-2Rs, expressed at low to intermediate levels on these cells, thus causing pulmonary edema. Notably, IL-2-mediated pulmonary edema was abrogated by a blocking antibody to IL-2Ralpha (CD25), genetic disruption of CD25, or the use of IL-2Rbetagamma-directed IL-2/anti-IL-2 antibody complexes, thereby interfering with IL-2 binding to IL-2Ralphabetagamma(+) pulmonary endothelial cells. Moreover, IL-2/anti-IL-2 antibody complexes led to vigorous activation of IL-2Rbetagamma(+) effector immune cells, which generated a dramatic antitumor response. Thus, IL-2/anti-IL-2 antibody complexes might improve current strategies of IL-2-based tumor immunotherapy.
in vivo CD8+ T cell depletion
Shariff, H., et al. (2010). "Intermittent antibody-based combination therapy removes alloantibodies and achieves indefinite heart transplant survival in presensitized recipients" Transplantation 90(3): 270-278. PubMed
BACKGROUND: It is well established that primed/memory T cells play a critical role in heart transplant rejection. This contributes to the challenges faced in the transplant clinic because current treatments that are efficient in controlling naive T cell alloresponses have limited efficacy on primed T cell responders. METHODS: Fully MHC-mismatched heart transplantation was performed from BALB/c to C57BL/6 mice presensitized with BALB/c splenocytes 14 days pretransplantation. A combination therapy comprising CD70-, CD154-, and CD8-specific antibodies (Abs) was administered at day 0 and 4 posttransplantation with rapamycin on days 0 to 4. RESULTS: The Ab combination therapy extended heart transplant survival in presensitized recipients from median survival time 8 days (MST) to MST 78 days. A decrease in the number of splenic interferon-gamma-secreting cells measured by ELISpot assay was seen in the treated group compared with the untreated controls. However, graft-infiltrating CD8+ and CD4+ T cells persisted despite treatment and the number of intragraft CD4+ T cells increased at day 30 posttransplantation. When an additional “rescue therapy” comprising the same Abs was readministered at days 30, 60, and 90 posttransplantation, T cell infiltration was reduced and indefinite graft survival was observed. Furthermore, rescue therapy resulted in gradual decrease in titer and, by day 90 posttransplantation, the complete loss of the preexisting, donor-specific Abs. CONCLUSION: We conclude that our Ab combination therapy extends allograft survival in presensitized recipients. When combined with intermittent Ab-mediated rescue therapy, this results in indefinite allograft survival and a loss of the preexisting, donor-specific Abs from the circulation.
in vivo CD8+ T cell depletion
Kish, D. D., et al. (2009). "CD8 T cells producing IL-17 and IFN-gamma initiate the innate immune response required for responses to antigen skin challenge" J Immunol 182(10): 5949-5959. PubMed
Effector CD8 T cell recruitment into the skin in response to Ag challenge requires prior CXCL1/KC-directed neutrophil infiltration. Mechanisms inducing CXCL1 production and the dynamics of neutrophil-CD8 T cell interactions during elicitation of Ag-specific responses in the skin were investigated. CXCL1 and CXCL2/MIP-2 were produced within 3-6 h of Ag challenge at 10-fold higher levels in skin challenge sites of Ag-sensitized vs nonsensitized mice. In the challenge sites of sensitized mice this production decreased at 6-9 h postchallenge to near the levels observed in skin challenge sites of nonsensitized mice but rose to a second peak 12 h after challenge. The elevated early neutrophil chemoattractant production at 3-6 h after skin challenge of sensitized animals required both IFN-gamma and IL-17, produced by distinct populations of Ag-primed CD8 T cells in response to Ag challenge. Although induced by the Ag-primed CD8 T cells, the early CXCL1 and CXCL2 production was accompanied by neutrophil but not CD8 T cell infiltration into the skin Ag challenge site. Infiltration of the CD8 T cells into the challenge site was not observed until 18-24 h after challenge. These results demonstrate an intricate series of early interactions between Ag-specific and innate immune components that regulate the sequential infiltration of neutrophils and then effector T cells into the skin to mediate an immune response.