InVivoMAb anti-mouse CD28
Product Details
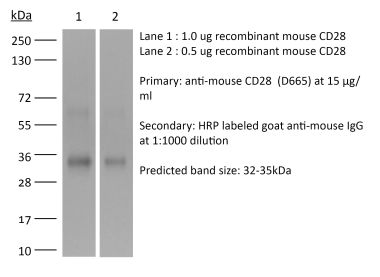
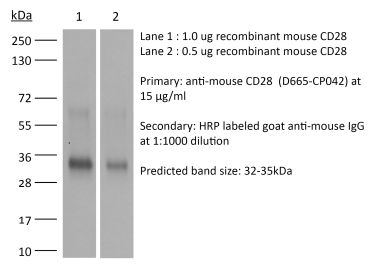
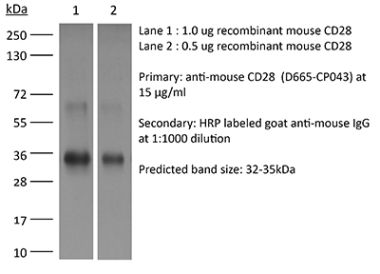
The 37.51 monoclonal antibody reacts with mouse CD28, a 45 kDa costimulatory receptor and a member of the Ig superfamily. CD28 is expressed by thymocytes, most peripheral T cells, and NK cells. CD28 is a receptor for CD80 (B7-1) and CD86 (B7-2). Signaling through CD28 augments IL-2 and IL-2 receptor expression as well as cytotoxicity of CD3-activated T cells. The 37.51 antibody has been shown to stimulate the proliferation and cytokine production by activated T and NK cells and provide a costimulatory signal for CTL induction.Specifications
| Isotype | Syrian Hamster IgG2 |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb polyclonal Syrian hamster IgG |
| Recommended Dilution Buffer | InVivoPure pH 6.0T Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | C57BL/6 mouse T cell lymphoma EL-4 cells |
| Reported Applications |
in vitro T cell stimulation/activation in vivo CD28 blockade |
| Formulation |
PBS, pH 6.0 0.01% Tween Contains no stabilizers or preservatives |
| Endotoxin |
<2EU/mg (<0.002EU/μg) Determined by LAL gel clotting assay |
| Purity |
>95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_1107624 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
Recommended Products
-
Recommended Isotype Control(s)
InVivoMAb polyclonal Syrian hamster IgG
-

Recommended Dilution Buffer
InVivoPure pH 6.0T Dilution Buffer
in vitro T cell stimulation/activation
Lacher, S. M., et al. (2018). "NF-kappaB inducing kinase (NIK) is an essential post-transcriptional regulator of T-cell activation affecting F-actin dynamics and TCR signaling" J Autoimmun 94: 110-121. PubMed
NF-kappaB inducing kinase (NIK) is the key protein of the non-canonical NF-kappaB pathway and is important for the development of lymph nodes and other secondary immune organs. We elucidated the specific role of NIK in T cells using T-cell specific NIK-deficient (NIK(DeltaT)) mice. Despite showing normal development of lymphoid organs, NIK(DeltaT) mice were resistant to induction of CNS autoimmunity. T cells from NIK(DeltaT) mice were deficient in late priming, failed to up-regulate T-bet and to transmigrate into the CNS. Proteomic analysis of activated NIK(-/-) T cells showed de-regulated expression of proteins involved in the formation of the immunological synapse: in particular, proteins involved in cytoskeleton dynamics. In line with this we found that NIK-deficient T cells were hampered in phosphorylation of Zap70, LAT, AKT, ERK1/2 and PLCgamma upon TCR engagement. Hence, our data disclose a hitherto unknown function of NIK in T-cell priming and differentiation.
in vitro T cell stimulation/activation
Wendland, K., et al. (2018). "Retinoic Acid Signaling in Thymic Epithelial Cells Regulates Thymopoiesis" J Immunol 201(2): 524-532. PubMed
Despite the essential role of thymic epithelial cells (TEC) in T cell development, the signals regulating TEC differentiation and homeostasis remain incompletely understood. In this study, we show a key in vivo role for the vitamin A metabolite, retinoic acid (RA), in TEC homeostasis. In the absence of RA signaling in TEC, cortical TEC (cTEC) and CD80(lo)MHC class II(lo) medullary TEC displayed subset-specific alterations in gene expression, which in cTEC included genes involved in epithelial proliferation, development, and differentiation. Mice whose TEC were unable to respond to RA showed increased cTEC proliferation, an accumulation of stem cell Ag-1(hi) cTEC, and, in early life, a decrease in medullary TEC numbers. These alterations resulted in reduced thymic cellularity in early life, a reduction in CD4 single-positive and CD8 single-positive numbers in both young and adult mice, and enhanced peripheral CD8(+) T cell survival upon TCR stimulation. Collectively, our results identify RA as a regulator of TEC homeostasis that is essential for TEC function and normal thymopoiesis.
in vitro T cell stimulation/activation
Ron-Harel, N., et al. (2016). "Mitochondrial Biogenesis and Proteome Remodeling Promote One-Carbon Metabolism for T Cell Activation" Cell Metab 24(1): 104-117. PubMed
Naive T cell stimulation activates anabolic metabolism to fuel the transition from quiescence to growth and proliferation. Here we show that naive CD4(+) T cell activation induces a unique program of mitochondrial biogenesis and remodeling. Using mass spectrometry, we quantified protein dynamics during T cell activation. We identified substantial remodeling of the mitochondrial proteome over the first 24 hr of T cell activation to generate mitochondria with a distinct metabolic signature, with one-carbon metabolism as the most induced pathway. Salvage pathways and mitochondrial one-carbon metabolism, fed by serine, contribute to purine and thymidine synthesis to enable T cell proliferation and survival. Genetic inhibition of the mitochondrial serine catabolic enzyme SHMT2 impaired T cell survival in culture and antigen-specific T cell abundance in vivo. Thus, during T cell activation, mitochondrial proteome remodeling generates specialized mitochondria with enhanced one-carbon metabolism that is critical for T cell activation and survival.
in vitro T cell stimulation/activation
Gu, A. D., et al. (2015). "A critical role for transcription factor Smad4 in T cell function that is independent of transforming growth factor beta receptor signaling" Immunity 42(1): 68-79. PubMed
Transforming growth factor-beta (TGF-beta) suppresses T cell function to maintain self-tolerance and to promote tumor immune evasion. Yet how Smad4, a transcription factor component of TGF-beta signaling, regulates T cell function remains unclear. Here we have demonstrated an essential role for Smad4 in promoting T cell function during autoimmunity and anti-tumor immunity. Smad4 deletion rescued the lethal autoimmunity resulting from transforming growth factor-beta receptor (TGF-betaR) deletion and compromised T-cell-mediated tumor rejection. Although Smad4 was dispensable for T cell generation, homeostasis, and effector function, it was essential for T cell proliferation after activation in vitro and in vivo. The transcription factor Myc was identified to mediate Smad4-controlled T cell proliferation. This study thus reveals a requirement of Smad4 for T-cell-mediated autoimmunity and tumor rejection, which is beyond the current paradigm. It highlights a TGF-betaR-independent role for Smad4 in promoting T cell function, autoimmunity, and anti-tumor immunity.
in vitro T cell stimulation/activation
Choi, Y. S., et al. (2015). "LEF-1 and TCF-1 orchestrate TFH differentiation by regulating differentiation circuits upstream of the transcriptional repressor Bcl6" Nat Immunol 16(9): 980-990. PubMed
Follicular helper T cells (TFH cells) are specialized effector CD4(+) T cells that help B cells develop germinal centers (GCs) and memory. However, the transcription factors that regulate the differentiation of TFH cells remain incompletely understood. Here we report that selective loss of Lef1 or Tcf7 (which encode the transcription factor LEF-1 or TCF-1, respectively) resulted in TFH cell defects, while deletion of both Lef1 and Tcf7 severely impaired the differentiation of TFH cells and the formation of GCs. Forced expression of LEF-1 enhanced TFH differentiation. LEF-1 and TCF-1 coordinated such differentiation by two general mechanisms. First, they established the responsiveness of naive CD4(+) T cells to TFH cell signals. Second, they promoted early TFH differentiation via the multipronged approach of sustaining expression of the cytokine receptors IL-6Ralpha and gp130, enhancing expression of the costimulatory receptor ICOS and promoting expression of the transcriptional repressor Bcl6.
in vitro T cell stimulation/activation
Xu, H., et al. (2015). "Regulation of bifurcating B cell trajectories by mutual antagonism between transcription factors IRF4 and IRF8" Nat Immunol . PubMed
Upon recognition of antigen, B cells undertake a bifurcated response in which some cells rapidly differentiate into plasmablasts while others undergo affinity maturation in germinal centers (GCs). Here we identified a double-negative feedback loop between the transcription factors IRF4 and IRF8 that regulated the initial developmental bifurcation of activated B cells as well as the GC response. IRF8 dampened signaling via the B cell antigen receptor (BCR), facilitated antigen-specific interaction with helper T cells, and promoted antibody affinity maturation while antagonizing IRF4-driven differentiation of plasmablasts. Genomic analysis revealed concentration-dependent actions of IRF4 and IRF8 in regulating distinct gene-expression programs. Stochastic modeling suggested that the double-negative feedback was sufficient to initiate bifurcation of the B cell developmental trajectories.
in vivo CD28 blockade
Rouhani, S. J., et al. (2015). "Roles of lymphatic endothelial cells expressing peripheral tissue antigens in CD4 T-cell tolerance induction" Nat Commun 6: 6771. PubMed
Lymphatic endothelial cells (LECs) directly express peripheral tissue antigens and induce CD8 T-cell deletional tolerance. LECs express MHC-II molecules, suggesting they might also tolerize CD4 T cells. We demonstrate that when beta-galactosidase (beta-gal) is expressed in LECs, beta-gal-specific CD8 T cells undergo deletion via the PD-1/PD-L1 and LAG-3/MHC-II pathways. In contrast, LECs do not present endogenous beta-gal in the context of MHC-II molecules to beta-gal-specific CD4 T cells. Lack of presentation is independent of antigen localization, as membrane-bound haemagglutinin and I-Ealpha are also not presented by MHC-II molecules. LECs express invariant chain and cathepsin L, but not H2-M, suggesting that they cannot load endogenous antigenic peptides onto MHC-II molecules. Importantly, LECs transfer beta-gal to dendritic cells, which subsequently present it to induce CD4 T-cell anergy. Therefore, LECs serve as an antigen reservoir for CD4 T-cell tolerance, and MHC-II molecules on LECs are used to induce CD8 T-cell tolerance via LAG-3.
in vitro T cell stimulation/activation
Rabenstein, H., et al. (2014). "Differential kinetics of antigen dependency of CD4+ and CD8+ T cells" J Immunol 192(8): 3507-3517. PubMed
Ag recognition via the TCR is necessary for the expansion of specific T cells that then contribute to adaptive immunity as effector and memory cells. Because CD4+ and CD8+ T cells differ in terms of their priming APCs and MHC ligands we compared their requirements of Ag persistence during their expansion phase side by side. Proliferation and effector differentiation of TCR transgenic and polyclonal mouse T cells were thus analyzed after transient and continuous TCR signals. Following equally strong stimulation, CD4+ T cell proliferation depended on prolonged Ag presence, whereas CD8+ T cells were able to divide and differentiate into effector cells despite discontinued Ag presentation. CD4+ T cell proliferation was neither affected by Th lineage or memory differentiation nor blocked by coinhibitory signals or missing inflammatory stimuli. Continued CD8+ T cell proliferation was truly independent of self-peptide/MHC-derived signals. The subset divergence was also illustrated by surprisingly broad transcriptional differences supporting a stronger propensity of CD8+ T cells to programmed expansion. These T cell data indicate an intrinsic difference between CD4+ and CD8+ T cells regarding the processing of TCR signals for proliferation. We also found that the presentation of a MHC class II-restricted peptide is more efficiently prolonged by dendritic cell activation in vivo than a class I bound one. In summary, our data demonstrate that CD4+ T cells require continuous stimulation for clonal expansion, whereas CD8+ T cells can divide following a much shorter TCR signal.
in vitro T cell stimulation/activation
Xiao, N., et al. (2014). "The E3 ubiquitin ligase Itch is required for the differentiation of follicular helper T cells" Nat Immunol 15(7): 657-666. PubMed
Follicular helper T cells (T(FH) cells) are responsible for effective B cell-mediated immunity, and Bcl-6 is a central factor for the differentiation of T(FH) cells. However, the molecular mechanisms that regulate the induction of T(FH) cells remain unclear. Here we found that the E3 ubiquitin ligase Itch was essential for the differentiation of T(FH) cells, germinal center responses and immunoglobulin G (IgG) responses to acute viral infection. Itch acted intrinsically in CD4(+) T cells at early stages of T(FH) cell development. Itch seemed to act upstream of Bcl-6 expression, as Bcl-6 expression was substantially impaired in Itch(-/-) cells, and the differentiation of Itch(-/-) T cells into T(FH) cells was restored by enforced expression of Bcl-6. Itch associated with the transcription factor Foxo1 and promoted its ubiquitination and degradation. The defective T(FH) differentiation of Itch(-/-) T cells was rectified by deletion of Foxo1. Thus, our results indicate that Itch acts as an essential positive regulator in the differentiation of T(FH) cells.
in vitro T cell stimulation/activation
Tang, W., et al. (2014). "The oncoprotein and transcriptional regulator Bcl-3 governs plasticity and pathogenicity of autoimmune T cells" Immunity 41(4): 555-566. PubMed
Bcl-3 is an atypical member of the IkappaB family that modulates transcription in the nucleus via association with p50 (NF-kappaB1) or p52 (NF-kappaB2) homodimers. Despite evidence attesting to the overall physiologic importance of Bcl-3, little is known about its cell-specific functions or mechanisms. Here we demonstrate a T-cell-intrinsic function of Bcl-3 in autoimmunity. Bcl-3-deficient T cells failed to induce disease in T cell transfer-induced colitis and experimental autoimmune encephalomyelitis. The protection against disease correlated with a decrease in Th1 cells that produced the cytokines IFN-gamma and GM-CSF and an increase in Th17 cells. Although differentiation into Th1 cells was not impaired in the absence of Bcl-3, differentiated Th1 cells converted to less-pathogenic Th17-like cells, in part via mechanisms involving expression of the RORgammat transcription factor. Thus, Bcl-3 constrained Th1 cell plasticity and promoted pathogenicity by blocking conversion to Th17-like cells, revealing a unique type of regulation that shapes adaptive immunity.
in vitro T cell stimulation/activation
Choi, Y. S., et al. (2013). "Bcl6 expressing follicular helper CD4 T cells are fate committed early and have the capacity to form memory" J Immunol 190(8): 4014-4026. PubMed
Follicular helper CD4 T (Tfh) cells are a distinct type of differentiated CD4 T cells uniquely specialized for B cell help. In this study, we examined Tfh cell fate commitment, including distinguishing features of Tfh versus Th1 proliferation and survival. Using cell transfer approaches at early time points after an acute viral infection, we demonstrate that early Tfh cells and Th1 cells are already strongly cell fate committed by day 3. Nevertheless, Tfh cell proliferation was tightly regulated in a TCR-dependent manner. The Tfh cells still depend on extrinsic cell fate cues from B cells in their physiological in vivo environment. Unexpectedly, we found that Tfh cells share a number of phenotypic parallels with memory precursor CD8 T cells, including selective upregulation of IL-7Ralpha and a collection of coregulated genes. As a consequence, the early Tfh cells can progress to robustly form memory cells. These data support the hypothesis that CD4 and CD8 T cells share core aspects of a memory cell precursor gene expression program involving Bcl6, and a strong relationship exists between Tfh cells and memory CD4 T cell development.
in vivo CD28 blockade
Eberlein, J., et al. (2012). "Multiple layers of CD80/86-dependent costimulatory activity regulate primary, memory, and secondary lymphocytic choriomeningitis virus-specific T cell immunity" J Virol 86(4): 1955-1970. PubMed
The lymphocytic choriomeningitis virus (LCMV) system constitutes one of the most widely used models for the study of infectious disease and the regulation of virus-specific T cell immunity. However, with respect to the activity of costimulatory and associated regulatory pathways, LCMV-specific T cell responses have long been regarded as relatively independent and thus distinct from the regulation of T cell immunity directed against many other viral pathogens. Here, we have reevaluated the contribution of CD28-CD80/86 costimulation in the LCMV system by use of CD80/86-deficient mice, and our results demonstrate that a disruption of CD28-CD80/86 signaling compromises the magnitude, phenotype, and/or functionality of LCMV-specific CD8(+) and/or CD4(+) T cell populations in all stages of the T cell response. Notably, a profound inhibition of secondary T cell immunity in LCMV-immune CD80/86-deficient mice emerged as a composite of both defective memory T cell development and a specific requirement for CD80 but not CD86 in the recall response, while a related experimental scenario of CD28-dependent yet CD80/86-independent secondary CD8(+) T cell immunity suggests the existence of a CD28 ligand other than CD80/86. Furthermore, we provide evidence that regulatory T cells (T(REG)s), the homeostasis of which is altered in CD80/86(-/-) mice, contribute to restrained LCMV-specific CD8(+) T cell responses in the presence of CD80/86. Our observations can therefore provide a more coherent perspective on CD28-CD80/86 costimulation in antiviral T cell immunity that positions the LCMV system within a shared context of multiple defects that virus-specific T cells acquire in the absence of CD28-CD80/86 costimulation.
in vitro T cell stimulation/activation
Angkasekwinai, P., et al. (2010). "Regulation of IL-9 expression by IL-25 signaling" Nat Immunol 11(3): 250-256. PubMed
The physiological regulation of the expression of interleukin (IL)-9, a cytokine traditionally regarded as being T(H)2 associated, remains unclear. Here, we show that IL-9-expressing T cells generated in vitro in the presence of transforming growth factor-beta and IL-4 express high levels of mRNA for IL-17 receptor B (IL-17RB), the receptor for IL-25. Treatment of these cells with IL-25 enhances IL-9 expression in vitro. Moreover, transgenic and retroviral overexpression of IL-17RB in T cells results in IL-25-induced IL-9 production that is IL-4 independent. In vivo, the IL-25-IL-17RB pathway regulates IL-9 expression in allergic airway inflammation. Thus, IL-25 is a newly identified regulator of IL-9 expression.