InVivoPlus anti-mouse OX40 (CD134)
Product Details
The OX-86 monoclonal antibody reacts with mouse OX-40 also known as CD134. OX-40 is a 50 kDa type I membrane glycoprotein and a member of the TNF receptor superfamily. OX-40 is expressed on activated CD4 and CD8 T cells, but is not found on resting naïve T cells or most resting memory T cells. Although it was originally thought that OX-40 expression was restricted to activated conventional T cells, it has now been visualized on activated regulatory T cells, NKT cells, NK cells, and neutrophils. OX-40 plays a major role in regulating both CD4 and CD8 T cell clonal expansion. It provides a costimulatory signal to an antigen-reacting naive T cells to prolong proliferation, as well as augment the production of several cytokines. This is demonstrated by OX-40 knockout mice which generate fewer primary effector CD4 T cells after immunization. Furthermore, in vivo treatment with an agonist antibody to OX-40 has been shown to strongly enhance the generation of antigen-specific effector T cells and prevent the induction of T cell tolerance. The OX-86 antibody is an agonistic antibody that has been shown to delay tumor growth in vivo.Specifications
| Isotype | Rat IgG1, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoPlus rat IgG1 isotype control, anti-horseradish peroxidase |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Recombinant mouse OX40-CD4 chimeric protein |
| Reported Applications |
in vivo OX40 activation in vitro OX40 activation Western blot |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin* |
<1EU/mg (<0.001EU/μg) Determined by LAL gel clotting assay |
| Aggregation* |
<5% Determined by SEC |
| Purity |
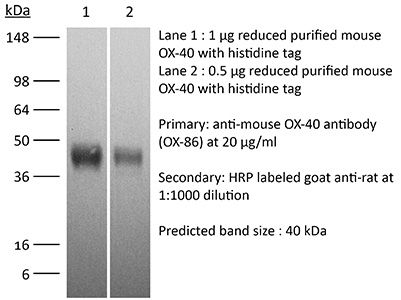
>95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_1107592 |
| Molecular Weight | 150 kDa |
| Murine Pathogen Tests* |
Ectromelia/Mousepox Virus: Negative Hantavirus: Negative K Virus: Negative Lactate Dehydrogenase-Elevating Virus: Negative Lymphocytic Choriomeningitis virus: Negative Mouse Adenovirus: Negative Mouse Cytomegalovirus: Negative Mouse Hepatitis Virus: Negative Mouse Minute Virus: Negative Mouse Norovirus: Negative Mouse Parvovirus: Negative Mouse Rotavirus: Negative Mycoplasma Pulmonis: Negative Pneumonia Virus of Mice: Negative Polyoma Virus: Negative Reovirus Screen: Negative Sendai Virus: Negative Theiler’s Murine Encephalomyelitis: Negative |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
Additional Formats

Recommended Products
-

Recommended Isotype Control(s)
InVivoPlus rat IgG1 isotype control, anti-horseradish peroxidase
-

Recommended Dilution Buffer
InVivoPure pH 7.0 Dilution Buffer
in vivo OX40 activation
Zander, R. A., et al. (2015). "PD-1 Co-inhibitory and OX40 Co-stimulatory Crosstalk Regulates Helper T Cell Differentiation and Anti-Plasmodium Humoral Immunity" Cell Host Microbe 17(5): 628-641. PubMed
The differentiation and protective capacity of Plasmodium-specific T cells are regulated by both positive and negative signals during malaria, but the molecular and cellular details remain poorly defined. Here we show that malaria patients and Plasmodium-infected rodents exhibit atypical expression of the co-stimulatory receptor OX40 on CD4 T cells and that therapeutic enhancement of OX40 signaling enhances helper CD4 T cell activity, humoral immunity, and parasite clearance in rodents. However, these beneficial effects of OX40 signaling are abrogated following coordinate blockade of PD-1 co-inhibitory pathways, which are also upregulated during malaria and associated with elevated parasitemia. Co-administration of biologics blocking PD-1 and promoting OX40 signaling induces excessive interferon-gamma that directly limits helper T cell-mediated support of humoral immunity and decreases parasite control. Our results show that targeting OX40 can enhance Plasmodium control and that crosstalk between co-inhibitory and co-stimulatory pathways in pathogen-specific CD4 T cells can impact pathogen clearance.
in vivo OX40 activation
Makkouk, A., et al. (2015). "Three steps to breaking immune tolerance to lymphoma: a microparticle approach" Cancer Immunol Res 3(4): 389-398. PubMed
In situ immunization aims at generating antitumor immune responses through manipulating the tumor microenvironment. On the basis of recent advances in the understanding of antitumor immunity, we designed a three-step approach to in situ immunization to lymphoma: (i) inducing immunogenic tumor cell death with the chemotherapeutic drug doxorubicin. Doxorubicin enhances the expression of “eat-me” signals by dying tumor cells, facilitating their phagocytosis by dendritic cells (DC). Because of the vesicant activity of doxorubicin, microparticles made of biodegradable polymer poly(lactide-co-glycolide) or PLGA can safely deliver doxorubicin intratumorally and are effective vaccine adjuvants, (ii) enhancing T-cell activation using anti-OX40 and (iii) sustaining T-cell responses by checkpoint blockade using anti-CTLA-4. In vitro, doxorubicin microparticles were less cytotoxic to DCs than to B lymphoma cells, did not require internalization by tumor cells, and significantly enhanced phagocytosis of tumor cells by DCs as compared with soluble doxorubicin. In mice, this three-step therapy induced CD4- and CD8-dependent systemic immune responses that enhanced T-cell infiltration into distant tumors, leading to their eradication and significantly improving survival. Our findings demonstrate that systemic antitumor immune responses can be generated locally by three-step therapy and merit further investigation as an immunotherapy for patients with lymphoma.
in vivo OX40 activation
Bartkowiak, T., et al. (2015). "Unique potential of 4-1BB agonist antibody to promote durable regression of HPV+ tumors when combined with an E6/E7 peptide vaccine" Proc Natl Acad Sci U S A 112(38): E5290-5299. PubMed
Antibody modulation of T-cell coinhibitory (e.g., CTLA-4) or costimulatory (e.g., 4-1BB) receptors promotes clinical responses to a variety of cancers. Therapeutic cancer vaccination, in contrast, has produced limited clinical benefit and no curative therapies. The E6 and E7 oncoproteins of human papilloma virus (HPV) drive the majority of genital cancers, and many oropharyngeal tumors. We discovered 15-19 amino acid peptides from HPV-16 E6/E7 for which induction of T-cell immunity correlates with disease-free survival in patients treated for high-grade cervical neoplasia. We report here that intranasal vaccination with these peptides and the adjuvant alpha-galactosylceramide elicits systemic and mucosal T-cell responses leading to reduced HPV(+) TC-1 tumor growth and prolonged survival in mice. We hypothesized that the inability of these T cells to fully reject established tumors resulted from suppression in the tumor microenvironment which could be ameliorated through checkpoint modulation. Combining this E6/E7 peptide vaccine with checkpoint blockade produced only modest benefit; however, coadministration with a 4-1BB agonist antibody promoted durable regression of established genital TC-1 tumors. Relative to other therapies tested, this combination of vaccine and alpha4-1BB promoted the highest CD8(+) versus regulatory FoxP3(+) T-cell ratios, elicited 2- to 5-fold higher infiltration by E7-specific CTL, and evoked higher densities of highly cytotoxic TcEO (T cytotoxic Eomesodermin) CD8 (>70-fold) and ThEO (T helper Eomesodermin) CD4 (>17-fold) T cells. These findings have immediate clinical relevance both in terms of the direct clinical utility of the vaccine studied and in illustrating the potential of 4-1BB antibody to convert therapeutic E6/E7 vaccines already in clinical trials into curative therapies.
in vivo OX40 activation
Krupnick, A. S., et al. (2014). "Central memory CD8+ T lymphocytes mediate lung allograft acceptance" J Clin Invest 124(3): 1130-1143. PubMed
Memory T lymphocytes are commonly viewed as a major barrier for long-term survival of organ allografts and are thought to accelerate rejection responses due to their rapid infiltration into allografts, low threshold for activation, and ability to produce inflammatory mediators. Because memory T cells are usually associated with rejection, preclinical protocols have been developed to target this population in transplant recipients. Here, using a murine model, we found that costimulatory blockade-mediated lung allograft acceptance depended on the rapid infiltration of the graft by central memory CD8+ T cells (CD44(hi)CD62L(hi)CCR7+). Chemokine receptor signaling and alloantigen recognition were required for trafficking of these memory T cells to lung allografts. Intravital 2-photon imaging revealed that CCR7 expression on CD8+ T cells was critical for formation of stable synapses with antigen-presenting cells, resulting in IFN-gamma production, which induced NO and downregulated alloimmune responses. Thus, we describe a critical role for CD8+ central memory T cells in lung allograft acceptance and highlight the need for tailored approaches for tolerance induction in the lung.
in vivo OX40 activation
Redmond, W. L., et al. (2014). "Combined targeting of costimulatory (OX40) and coinhibitory (CTLA-4) pathways elicits potent effector T cells capable of driving robust antitumor immunity" Cancer Immunol Res 2(2): 142-153. PubMed
Ligation of the TNF receptor family costimulatory molecule OX40 (CD134) with an agonist anti-OX40 monoclonal antibody (mAb) enhances antitumor immunity by augmenting T-cell differentiation as well as turning off the suppressive activity of the FoxP3(+)CD4(+) regulatory T cells (Treg). In addition, antibody-mediated blockade of the checkpoint inhibitor CTLA-4 releases the “brakes” on T cells to augment tumor immunotherapy. However, monotherapy with these agents has limited therapeutic benefit particularly against poorly immunogenic murine tumors. Therefore, we examined whether the administration of agonist anti-OX40 therapy in the presence of CTLA-4 blockade would enhance tumor immunotherapy. Combined anti-OX40/anti-CTLA-4 immunotherapy significantly enhanced tumor regression and the survival of tumor-bearing hosts in a CD4 and CD8 T cell-dependent manner. Mechanistic studies revealed that the combination immunotherapy directed the expansion of effector T-bet(high)/Eomes(high) granzyme B(+) CD8 T cells. Dual immunotherapy also induced distinct populations of Th1 [interleukin (IL)-2, IFN-gamma], and, surprisingly, Th2 (IL-4, IL-5, and IL-13) CD4 T cells exhibiting increased T-bet and Gata-3 expression. Furthermore, IL-4 blockade inhibited the Th2 response, while maintaining the Th1 CD4 and effector CD8 T cells that enhanced tumor-free survival. These data demonstrate that refining the global T-cell response during combination immunotherapy can further enhance the therapeutic efficacy of these agents.
in vivo OX40 activation
Guo, Z., et al. (2014). "PD-1 blockade and OX40 triggering synergistically protects against tumor growth in a murine model of ovarian cancer" PLoS One 9(2): e89350. PubMed
The co-inhibitory receptor Programmed Death-1 (PD-1) curtails immune responses and prevent autoimmunity, however, tumors exploit this pathway to escape from immune destruction. The co-stimulatory receptor OX40 is upregulated on T cells following activation and increases their clonal expansion, survival and cytokine production when engaged. Although antagonistic anti-PD-1 or agonistic anti-OX40 antibodies can promote the rejection of several murine tumors, some poorly immunogenic tumors were refractory to this treatment. In the present study, we evaluated the antitumor effects and mechanisms of combinatorial PD-1 blockade and OX40 triggering in a murine ID8 ovarian cancer model. Although individual anti-PD-1 or OX40 mAb treatment was ineffective in tumor protection against 10-day established ID8 tumor, combined anti-PD-1/OX40 mAb treatment markedly inhibited tumor outgrowth with 60% of mice tumor free 90 days after tumor inoculation. Tumor protection was associated with a systemic immune response with memory and antigen specificity and required CD4(+) cells and CD8(+) T cells. The anti-PD-1/OX40 mAb treatment increased CD4(+) and CD8(+) cells and decreased immunosuppressive CD4(+)FoxP3(+) regulatory T (Treg) cells and CD11b(+)Gr-1(+) myeloid suppressor cells (MDSC), giving rise to significantly higher ratios of both effector CD4(+) and CD8(+) cells to Treg and MDSC in peritoneal cavity; Quantitative RT-PCR data further demonstrated the induction of a local immunostimulatory milieu by anti-PD-1/OX40 mAb treatment. The splenic CD8(+) T cells from combined mAb treated mice produced high levels of IFN-gamma upon tumor antigen stimulation and exhibited antigen-specific cytolytic activity. To our knowledge, this is the first study testing the antitumor effects of combined anti-PD-1/OX40 mAb in a murine ovarian cancer model, and our results provide a rationale for clinical trials evaluating ovarian cancer immunotherapy using this combination of mAb.
in vitro OX40 activation
Hu, Z., et al. (2013). "Regulatory CD8+ T cells associated with erosion of immune surveillance in persistent virus infection suppress in vitro and have a reversible proliferative defect" J Immunol 191(1): 312-322. PubMed
CD4(+) T cell help is critical for CD8(+) T cell memory and immune surveillance against persistent virus infections. Our recent data have showed the lack of CD4(+) T cells leads to the generation of an IL-10-producing CD8(+) T cell population during persistent murine gamma-herpesvirus-68 (MHV-68) infection. IL-10 from these cells is partly responsible for erosion in immune surveillance, leading to spontaneous virus reactivation in lungs. In this study, we further characterized the generation, phenotype, and function of these IL-10-producing CD8(+) T cells by comparing with a newly identified IL-10-producing CD8(+) T cell population present during the acute stage of the infection. The IL-10-producing CD8(+) populations in acute and chronic stages differed in their requirement for CD4(+) T cell help, the dependence on IL-2/CD25 and CD40-CD40L pathways, and the ability to proliferate in vitro in response to anti-CD3 stimulation. IL-10-producing CD8(+) T cells in the chronic stage showed a distinct immunophenotypic profile, sharing partial overlap with the markers of previously reported regulatory CD8(+) T cells, and suppressed the proliferation of naive CD8(+) T cells. Notably, they retained the ability to produce effector cytokines and cytotoxic activity. In addition, the proliferative defect of the cells could be restored by addition of exogenous IL-2 or blockade of IL-10. These data suggest that the IL-10-producing CD8(+) T cells arising in chronic MHV-68 infection in the absence of CD4(+) T cell help belong to a subset of CD8(+) regulatory T cells.
in vivo OX40 activation
Xiao, X., et al. (2012). "New insights on OX40 in the control of T cell immunity and immune tolerance in vivo" J Immunol 188(2): 892-901. PubMed
OX40 is a T cell costimulatory molecule that belongs to the TNFR superfamily. In the absence of immune activation, OX40 is selectively expressed by Foxp3(+) regulatory T cells (Tregs), but not by resting conventional T cells. The exact role of OX40 in Treg homeostasis and function remains incompletely defined. In this study, we demonstrate that OX40 engagement in vivo in naive mice induces initial expansion of Foxp3(+) Tregs, but the expanded Tregs have poor suppressive function and exhibit features of exhaustion. We also show that OX40 enables the activation of the Akt and Stat5 pathways in Tregs, resulting in transient proliferation of Tregs and reduced levels of Foxp3 expression. This creates a state of relative IL-2 deficiency in naive mice that further impacts Tregs. This exhausted Treg phenotype can be prevented by exogenous IL-2, as both OX40 and IL-2 agonists drive further expansion of Tregs in vivo. Importantly, Tregs expanded by both OX40 and IL-2 agonists are potent suppressor cells, and in a heart transplant model, they promote long-term allograft survival. Our data reveal a novel role for OX40 in promoting immune tolerance and may have important clinical implications.
in vivo OX40 activation
Kurche, J. S., et al. (2012). "Type I IFN-dependent T cell activation is mediated by IFN-dependent dendritic cell OX40 ligand expression and is independent of T cell IFNR expression" J Immunol 188(2): 585-593. PubMed
Type I IFNs are important for direct control of viral infection and generation of adaptive immune responses. Recently, direct stimulation of CD4(+) T cells via type I IFNR has been shown to be necessary for the formation of functional CD4(+) T cell responses. In contrast, we find that CD4(+) T cells do not require intrinsic type I IFN signals in response to combined TLR/anti-CD40 vaccination. Rather, the CD4 response is dependent on the expression of type I IFNR (IFNalphaR) on innate cells. Further, we find that dendritic cell (DC) expression of the TNF superfamily member OX40 ligand was dependent on type I IFN signaling in the DC, resulting in a reduced CD4(+) T cell response that could be substantially rescued by an agonistic Ab to the receptor OX40. Taken together, we show that the IFNalphaR dependence of the CD4(+) T cell response is accounted for exclusively by defects in DC activation.
in vivo OX40 activation
Murray, S. E., et al. (2011). "NF-kappaB-inducing kinase plays an essential T cell-intrinsic role in graft-versus-host disease and lethal autoimmunity in mice" J Clin Invest 121(12): 4775-4786. PubMed
NF-kappaB-inducing kinase (NIK) is an essential upstream kinase in noncanonical NF-kappaB signaling. NIK-dependent NF-kappaB activation downstream of several TNF receptor family members mediates lymphoid organ development and B cell homeostasis. Peripheral T cell populations are normal in the absence of NIK, but the role of NIK during in vivo T cell responses to antigen has been obscured by other developmental defects in NIK-deficient mice. Here, we have identified a T cell-intrinsic requirement for NIK in graft-versus-host disease (GVHD), wherein NIK-deficient mouse T cells transferred into MHC class II mismatched recipients failed to cause GVHD. Although NIK was not necessary for antigen receptor signaling, it was absolutely required for costimulation through the TNF receptor family member OX40 (also known as CD134). When we conditionally overexpressed NIK in T cells, mice suffered rapid and fatal autoimmunity characterized by hyperactive effector T cells and poorly suppressive Foxp3(+) Tregs. Together, these data illuminate a critical T cell-intrinsic role for NIK during immune responses and suggest that its tight regulation is critical for avoiding autoimmunity.