InVivoPlus anti-mouse 4-1BB (CD137)

Product Details
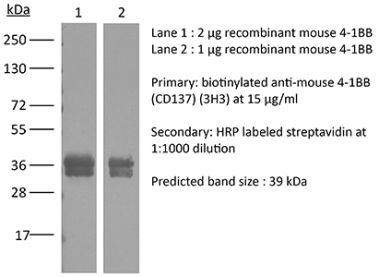
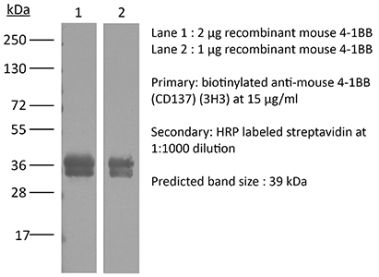
The LOB12.3 monoclonal antibody reacts with mouse 4-1BB, a TNF receptor superfamily member also known as CD137. 4-1BB is a 39 kDa transmembrane protein expressed by T lymphocytes, NK cells, dendritic cells, granulocytes, and mast cells. Upon binding its ligand 4-1BBL, 4-1BB provides costimulatory signals to both CD4 and CD8 T cells through the activation of NF- κB, c-Jun and p38 downstream pathways. The importance of the 4-1BB pathway has been underscored in a number of diseases, including cancer. Agonistic anti-4-1BB antibodies have been reported to induce T cell mediated antitumor immunity. The LOB12.3 antibody is an agonistic antibody that has been shown to stimulate 4-1BB signaling and delay tumor growth in vivo when administered in combination with immune checkpoint inhibitors.Specifications
| Isotype | Rat IgG1, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoPlus rat IgG1 isotype control, anti-horseradish peroxidase |
| Recommended Dilution Buffer | InVivoPure pH 8.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Mouse CD137 human Fc fusion protein |
| Reported Applications | in vivo activation of 4-1BB |
| Formulation |
PBS, pH 8.0 Contains no stabilizers or preservatives |
| Endotoxin* |
<1EU/mg (<0.001EU/μg) Determined by LAL gel clotting assay |
| Aggregation* |
<5% Determined by SEC |
| Purity |
>95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_10949016 |
| Molecular Weight | 150 kDa |
| Murine Pathogen Tests* |
Ectromelia/Mousepox Virus: Negative Hantavirus: Negative K Virus: Negative Lactate Dehydrogenase-Elevating Virus: Negative Lymphocytic Choriomeningitis virus: Negative Mouse Adenovirus: Negative Mouse Cytomegalovirus: Negative Mouse Hepatitis Virus: Negative Mouse Minute Virus: Negative Mouse Norovirus: Negative Mouse Parvovirus: Negative Mouse Rotavirus: Negative Mycoplasma Pulmonis: Negative Pneumonia Virus of Mice: Negative Polyoma Virus: Negative Reovirus Screen: Negative Sendai Virus: Negative Theiler’s Murine Encephalomyelitis: Negative |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
Additional Formats


Recommended Products
-

Recommended Isotype Control(s)
InVivoPlus rat IgG1 isotype control, anti-horseradish peroxidase
-

Recommended Dilution Buffer
InVivoPure pH 8.0 Dilution Buffer
in vivo activation of 4-1BB
Qi, X., et al. (2019). "Optimization of 4-1BB antibody for cancer immunotherapy by balancing agonistic strength with FcgammaR affinity" Nat Commun 10(1): 2141. PubMed
Costimulation of T cell responses with monoclonal antibody agonists (mAb-AG) targeting 4-1BB showed robust anti-tumor activity in preclinical models, but their clinical development was hampered by low efficacy (Utomilumab) or severe liver toxicity (Urelumab). Here we show that isotype and intrinsic agonistic strength co-determine the efficacy and toxicity of anti-4-1BB mAb-AG. While intrinsically strong agonistic anti-4-1BB can activate 4-1BB in the absence of FcgammaRs, weak agonistic antibodies rely on FcgammaRs to activate 4-1BB. All FcgammaRs can crosslink anti-41BB antibodies to strengthen co-stimulation, but activating FcgammaR-induced antibody-dependent cell-mediated cytotoxicity compromises anti-tumor immunity by deleting 4-1BB(+) cells. This suggests balancing agonistic activity with the strength of FcgammaR interaction as a strategy to engineer 4-1BB mAb-AG with optimal therapeutic performance. As a proof of this concept, we have developed LVGN6051, a humanized 4-1BB mAb-AG that shows high anti-tumor efficacy in the absence of liver toxicity in a mouse model of cancer immunotherapy.
in vivo activation of 4-1BB
Dai, M., et al. (2015). "Curing mice with large tumors by locally delivering combinations of immunomodulatory antibodies" Clin Cancer Res 21(5): 1127-1138. PubMed
PURPOSE: Immunomodulatory mAbs can treat cancer, but cures are rare except for small tumors. Our objective was to explore whether the therapeutic window increases by combining mAbs with different modes of action and injecting them into tumors. EXPERIMENTAL DESIGN: Combinations of mAbs to CD137/PD-1/CTLA-4 or CD137/PD-1/CTLA-4/CD19 were administrated intratumorally to mice with syngeneic tumors (B16 and SW1 melanoma, TC1 lung carcinoma), including tumors with a mean surface of approximately 80 mm(2). Survival and tumor growth were assessed. Immunologic responses were evaluated using flow cytometry and qRT-PCR. RESULTS: More than 50% of tumor-bearing mice had complete regression and long-term survival after tumor injection with mAbs recognizing CD137/PD-1/CTLA-4/CD19 with similar responses in three models. Intratumoral injection was more efficacious than intraperitoneal injection in causing rejection also of untreated tumors in the same mice. The three-mAb combination could also induce regression, but was less efficacious. There were few side effects, and therapy-resistant tumors were not observed. Transplanted tumor cells rapidly caused a Th2 response with increased CD19 cells. Successful therapy shifted this response to the Th1 phenotype with decreased CD19 cells and increased numbers of long-term memory CD8 effector cells and T cells making IFNgamma and TNFalpha. CONCLUSIONS: Intratumoral injection of mAbs recognizing CD137/PD-1/CTLA-4/CD19 can eradicate established tumors and reverse a Th2 response with tumor-associated CD19 cells to Th1 immunity, whereas a combination lacking anti-CD19 is less effective. There are several human cancers for which a similar approach may provide clinical benefit.
in vivo activation of 4-1BB
Bartkowiak, T., et al. (2015). "Unique potential of 4-1BB agonist antibody to promote durable regression of HPV+ tumors when combined with an E6/E7 peptide vaccine" Proc Natl Acad Sci U S A 112(38): E5290-5299. PubMed
Antibody modulation of T-cell coinhibitory (e.g., CTLA-4) or costimulatory (e.g., 4-1BB) receptors promotes clinical responses to a variety of cancers. Therapeutic cancer vaccination, in contrast, has produced limited clinical benefit and no curative therapies. The E6 and E7 oncoproteins of human papilloma virus (HPV) drive the majority of genital cancers, and many oropharyngeal tumors. We discovered 15-19 amino acid peptides from HPV-16 E6/E7 for which induction of T-cell immunity correlates with disease-free survival in patients treated for high-grade cervical neoplasia. We report here that intranasal vaccination with these peptides and the adjuvant alpha-galactosylceramide elicits systemic and mucosal T-cell responses leading to reduced HPV(+) TC-1 tumor growth and prolonged survival in mice. We hypothesized that the inability of these T cells to fully reject established tumors resulted from suppression in the tumor microenvironment which could be ameliorated through checkpoint modulation. Combining this E6/E7 peptide vaccine with checkpoint blockade produced only modest benefit; however, coadministration with a 4-1BB agonist antibody promoted durable regression of established genital TC-1 tumors. Relative to other therapies tested, this combination of vaccine and alpha4-1BB promoted the highest CD8(+) versus regulatory FoxP3(+) T-cell ratios, elicited 2- to 5-fold higher infiltration by E7-specific CTL, and evoked higher densities of highly cytotoxic TcEO (T cytotoxic Eomesodermin) CD8 (>70-fold) and ThEO (T helper Eomesodermin) CD4 (>17-fold) T cells. These findings have immediate clinical relevance both in terms of the direct clinical utility of the vaccine studied and in illustrating the potential of 4-1BB antibody to convert therapeutic E6/E7 vaccines already in clinical trials into curative therapies.
in vivo activation of 4-1BB
Murphy, J. T., et al. (2014). "Anaphylaxis caused by repetitive doses of a GITR agonist monoclonal antibody in mice" Blood 123(14): 2172-2180. PubMed
Immunotherapy for cancer using antibodies to enhance T-cell function has been successful in recent clinical trials. Many molecules that improve activation and effector function of T cells have been investigated as potential new targets for immunomodulatory antibodies, including the tumor necrosis factor receptor superfamily members GITR and OX40. Antibodies engaging GITR or OX40 result in significant tumor protection in preclinical models. In this study, we observed that the GITR agonist antibody DTA-1 causes anaphylaxis in mice upon repeated intraperitoneal dosing. DTA-1-induced anaphylaxis requires GITR, CD4(+) T cells, B cells, and interleukin-4. Transfer of serum antibodies from DTA-1-treated mice, which contain high levels of DTA-1-specific immunoglobulin G1 (IgG1), can induce anaphylaxis in naive mice upon administration of an additional dose of DTA-1, suggesting that anaphylaxis results from anti-DTA-1 antibodies. Depletion of basophils and blockade of platelet-activating factor, the key components of the IgG1 pathway of anaphylaxis, rescues the mice from DTA-1-induced anaphylaxis. These results demonstrate a previously undescribed lethal side effect of repetitive doses of an agonist immunomodulatory antibody as well as insight into the mechanism of toxicity, which may offer a means of preventing adverse effects in future clinical trials using anti-GITR or other agonist antibodies as immunotherapies.
in vivo activation of 4-1BB
Manzke, N., et al. (2013). "CD4+ T cells develop antiretroviral cytotoxic activity in the absence of regulatory T cells and CD8+ T cells" J Virol 87(11): 6306-6313. PubMed
Conventional CD4(+) T cells play an important role in viral immunity. In most virus infections, they provide essential help for antiviral B and T cell responses. In chronic infections, including HIV infection, an expansion of regulatory T cells (Tregs) has been demonstrated, which can suppress virus-specific CD4(+) T cell responses in vitro. However, the suppressive activity of Tregs on effector CD4(+) T cells in retroviral infection is less well documented in vivo. We took advantage of a transgenic mouse in which Tregs can be selectively depleted to determine the influence of such cells on retrovirus-specific CD4(+) T cell responses during an ongoing infection. Mice were infected with Friend retrovirus (FV), and Tregs were depleted during the acute phase of the infection. In nondepleted mice, activated CD4(+) T cells produced Th1-type cytokines but did not exhibit any antiviral cytotoxicity as determined in a major histocompatibility complex (MHC) class II-restricted in vivo cytotoxic T lymphocyte (CTL) assay. Depletion of Tregs significantly increased the numbers of virus-specific CD4(+) T cells and improved their cytokine production, whereas it induced only very little CD4(+) T cell cytotoxicity. However, after dual depletion of Tregs and CD8(+) T cells, conventional CD4(+) T cells developed significant cytotoxic activity against FV epitope-labeled target cells in vivo and contributed to the control of virus replication. Thus, both Tregs and CD8(+) T cells influence the cytotoxic activity of conventional CD4(+) T cells during an acute retroviral infection.
in vivo activation of 4-1BB
Wei, H., et al. (2013). "Combinatorial PD-1 blockade and CD137 activation has therapeutic efficacy in murine cancer models and synergizes with cisplatin" PLoS One 8(12): e84927. PubMed
90 days (and was probably curative) by a mechanism which included a systemic CD8(+) T cell response with tumor specificity and immunological memory. Strikingly, combined treatment of cisplatin and CD137/PD-1 mAb also gave rise to the long-term survival of mice with established TC1 lung tumors. A similar combination of the 2 mAbs and cisplatin should be considered for clinical ‘translation’.”}” data-sheets-userformat=”{“2″:14851,”3”:{“1″:0},”4”:{“1″:2,”2″:16777215},”12″:0,”14”:{“1″:2,”2″:1521491},”15″:”Roboto, sans-serif”,”16″:12}”>There is an urgent need for improved therapy for advanced ovarian carcinoma, which may be met by administering immune-modulatory monoclonal antibodies (mAbs) to generate a tumor-destructive immune response. Using the ID8 mouse ovarian cancer model, we investigated the therapeutic efficacy of various mAb combinations in mice with intraperitoneal (i.p.) tumor established by transplanting 3 x 10(6) ID8 cells 10 days previously. While most of the tested mAbs were ineffective when given individually or together, the data confirm our previous finding that 2 i.p. injections of a combination of anti-CD137 with anti-PD-1 mAbs doubles overall survival. Mice treated with this mAb combination have a significantly increased frequency and total number of CD8(+) T cells both in the peritoneal lavage and spleens, and these cells are functional as demonstrated by antigen-specific cytolytic activity and IFN-gamma production. While administration of anti-CD137 mAb as a single agent similarly increases CD8(+) T cells, these have no functional activity, which may be attributed to up-regulation of co-inhibitory PD-1 and TIM-3 molecules induced by CD137. Addition of the anti-cancer drug cisplatin to the 2 mAb combination increased overall survival >90 days (and was probably curative) by a mechanism which included a systemic CD8(+) T cell response with tumor specificity and immunological memory. Strikingly, combined treatment of cisplatin and CD137/PD-1 mAb also gave rise to the long-term survival of mice with established TC1 lung tumors. A similar combination of the 2 mAbs and cisplatin should be considered for clinical ‘translation’.
in vivo activation of 4-1BB
Guo, Z., et al. (2013). "Combined TIM-3 blockade and CD137 activation affords the long-term protection in a murine model of ovarian cancer" J Transl Med 11: 215. PubMed
BACKGROUND: T-cell immunoglobulin and mucin domain 3 (TIM-3) is known as a negative immune regulator and emerging data have implicated TIM-3 a pivotal role in suppressing antitumor immunity. The co-stimulatory receptor CD137 is transiently upregulated on T-cells following activation and increases their proliferation and survival when engaged. Although antagonistic anti-TIM-3 or agonistic anti-CD137 antibodies can promote the rejection of several murine tumors, some poorly immunogenic tumors were refractory to this treatment. In this study, we sought to evaluate whether combined TIM-3 blockade and CD137 activation would significantly improve the immunotherapy in the murine ID8 ovarian cancer model. METHODS: Mice with established ID8 tumor were intraperitoneally injected with single or combined anti-TIM-3/CD137 monoclonal antibody (mAb); mice survival was recorded, the composition and gene expression of tumor-infiltrating immune cells in these mice was analyzed by flow cytometry and quantitative RT-PCR respectively, and the function of CD8(+) cells was evaluated by ELISA and cytotoxicity assay. RESULTS: Either anti-TIM-3 or CD137 mAb alone, although effective in 3 days established tumor, was unable to prevent tumor progression in mice bearing 10 days established tumor, however, combined anti-TIM-3/CD137 mAb significantly inhibited the growth of these tumors with 60% of mice tumor free 90 days after tumor inoculation. Therapeutic efficacy was associated with a systemic immune response with memory and antigen specificity, required CD4(+) cells and CD8(+) cells. The 2 mAb combination increased CD4(+) and CD8(+) cells and decreased immunosuppressive CD4(+)FoxP3(+) regulatory T (Treg) cells and CD11b(+)Gr-1(+) myeloid suppressor cells (MDSC) at tumor sites, giving rise to significantly elevated ratios of CD4(+) and CD8(+) cells to Treg and MDSC; This is consistent with biasing local immune response towards an immunostimulatory Th1 type and is further supported by quantitative RT-PCR data showing the increased Th1-associated genes by anti-TIM-3/CD137 treatment. The increased CD8(+) T cells produced high level of IFN-gamma upon tumor antigen stimulation and displayed antigen-specific cytotoxic activity. CONCLUSIONS: To our knowledge, this is the first report investigating the effects of anti-TIM-3/CD137 combined mAb in a murine ovarian cancer model, and our results may aid the design of future trials for ovarian cancer immunotherapy.
in vivo activation of 4-1BB
Kwong, B., et al. (2013). "Localized immunotherapy via liposome-anchored Anti-CD137 + IL-2 prevents lethal toxicity and elicits local and systemic antitumor immunity" Cancer Res 73(5): 1547-1558. PubMed
Immunostimulatory agonists such as anti-CD137 and interleukin (IL)-2 have elicited potent antitumor immune responses in preclinical studies, but their clinical use is limited by inflammatory toxicities that result upon systemic administration. We hypothesized that by rigorously restricting the biodistribution of immunotherapeutic agents to a locally accessible lesion and draining lymph node(s), effective local and systemic antitumor immunity could be achieved in the absence of systemic toxicity. We anchored anti-CD137 and an engineered IL-2Fc fusion protein to the surfaces of PEGylated liposomes, whose physical size permitted dissemination in the tumor parenchyma and tumor-draining lymph nodes but blocked entry into the systemic circulation following intratumoral injection. In the B16F10 melanoma model, intratumoral liposome-coupled anti-CD137 + IL-2Fc therapy cured a majority of established primary tumors while avoiding the lethal inflammatory toxicities caused by equivalent intratumoral doses of soluble immunotherapy. Immunoliposome therapy induced protective antitumor memory and elicited systemic antitumor immunity that significantly inhibited the growth of simultaneously established distal tumors. Tumor inhibition was CD8(+) T-cell-dependent and was associated with increased CD8(+) T-cell infiltration in both treated and distal tumors, enhanced activation of tumor antigen-specific T cells in draining lymph nodes, and a reduction in regulatory T cells in treated tumors. These data suggest that local nanoparticle-anchored delivery of immuno-agonists represents a promising strategy to improve the therapeutic window and clinical applicability of highly potent but otherwise intolerable regimens of cancer immunotherapy. Cancer Res; 73(5); 1547-58. (c)2012 AACR.
in vivo activation of 4-1BB
Dai, M., et al. (2013). "Long-lasting complete regression of established mouse tumors by counteracting Th2 inflammation" J Immunother 36(4): 248-257. PubMed
40% of mice with SW1 tumors remained healthy >150 days after last treatment and are probably cured. Therapeutic efficacy was associated with a systemic immune response with memory and antigen specificity, required CD4 cells and involved CD8 cells and NK cells to a less extent. The 3 mAb combination significantly decreased CD19 cells at tumor sites, increased IFN-gamma and TNF-alpha producing CD4 and CD8 T cells and mature CD86 dendritic cells (DC), and it increased the ratios of effector CD4 and CD8 T cells to CD4Foxp3 regulatory T (Treg) cells and to CD11bGr-1 myeloid suppressor cells (MDSC). This is consistent with shifting the tumor microenvironment from an immunosuppressive Th2 to an immunostimulatory Th1 type and is further supported by PCR data. Adding an anti-CD19 mAb to the 3 mAb combination in the SW1 model further increased therapeutic efficacy. Data from ongoing experiments show that intratumoral injection of a combination of mAbs to CD137PD-1CTLA4CD19 can induce complete regression and dramatically prolong survival also in the TC1 carcinoma and B16 melanoma models, suggesting that the approach has general validity.”}” data-sheets-userformat=”{“2″:14851,”3”:{“1″:0},”4”:{“1″:2,”2″:16777215},”12″:0,”14”:{“1″:2,”2″:1521491},”15″:”Roboto, sans-serif”,”16″:12}”>Mice with intraperitoneal ID8 ovarian carcinoma or subcutaneous SW1 melanoma were injected with monoclonal antibodies (mAbs) to CD137PD-1CTLA4 7-15 days after tumor initiation. Survival of mice with ID8 tumors tripled and >40% of mice with SW1 tumors remained healthy >150 days after last treatment and are probably cured. Therapeutic efficacy was associated with a systemic immune response with memory and antigen specificity, required CD4 cells and involved CD8 cells and NK cells to a less extent. The 3 mAb combination significantly decreased CD19 cells at tumor sites, increased IFN-gamma and TNF-alpha producing CD4 and CD8 T cells and mature CD86 dendritic cells (DC), and it increased the ratios of effector CD4 and CD8 T cells to CD4Foxp3 regulatory T (Treg) cells and to CD11bGr-1 myeloid suppressor cells (MDSC). This is consistent with shifting the tumor microenvironment from an immunosuppressive Th2 to an immunostimulatory Th1 type and is further supported by PCR data. Adding an anti-CD19 mAb to the 3 mAb combination in the SW1 model further increased therapeutic efficacy. Data from ongoing experiments show that intratumoral injection of a combination of mAbs to CD137PD-1CTLA4CD19 can induce complete regression and dramatically prolong survival also in the TC1 carcinoma and B16 melanoma models, suggesting that the approach has general validity.
in vivo activation of 4-1BB
Curran, M. A., et al. (2011). "Combination CTLA-4 blockade and 4-1BB activation enhances tumor rejection by increasing T-cell infiltration, proliferation, and cytokine production" PLoS One 6(4): e19499. PubMed
BACKGROUND: The co-inhibitory receptor Cytotoxic T-Lymphocyte Antigen 4 (CTLA-4) attenuates immune responses and prevent autoimmunity, however, tumors exploit this pathway to evade the host T-cell response. The T-cell co-stimulatory receptor 4-1BB is transiently upregulated on T-cells following activation and increases their proliferation and inflammatory cytokine production when engaged. Antibodies which block CTLA-4 or which activate 4-1BB can promote the rejection of some murine tumors, but fail to cure poorly immunogenic tumors like B16 melanoma as single agents. METHODOLOGY/PRINCIPAL FINDINGS: We find that combining alphaCTLA-4 and alpha4-1BB antibodies in the context of a Flt3-ligand, but not a GM-CSF, based B16 melanoma vaccine promoted synergistic levels of tumor rejection. 4-1BB activation elicited strong infiltration of CD8+ T-cells into the tumor and drove the proliferation of these cells, while CTLA-4 blockade did the same for CD4+ effector T-cells. Anti-4-1BB also depressed regulatory T-cell infiltration of tumors. 4-1BB activation strongly stimulated inflammatory cytokine production in the vaccine and tumor draining lymph nodes and in the tumor itself. The addition of CTLA-4 blockade further increased IFN-gamma production from CD4+ effector T-cells in the vaccine draining node and the tumor. Anti 4-1BB treatment, with or without CTLA-4 blockade, induced approximately 75% of CD8+ and 45% of CD4+ effector T-cells in the tumor to express the killer cell lectin-like receptor G1 (KLRG1). Tumors treated with combination antibody therapy showed 1.7-fold greater infiltration by these KLRG1+CD4+ effector T-cells than did those treated with alpha4-1BB alone. CONCLUSIONS/SIGNIFICANCE: This study shows that combining T-cell co-inhibitory blockade with alphaCTLA-4 and active co-stimulation with alpha4-1BB promotes rejection of B16 melanoma in the context of a suitable vaccine. In addition, we identify KLRG1 as a useful marker for monitoring the anti-tumor immune response elicited by this therapy. These findings should aid in the design of future trials for the immunotherapy of melanoma.
in vivo activation of 4-1BB
Taraban, V. Y., et al. (2002). "Expression and costimulatory effects of the TNF receptor superfamily members CD134 (OX40) and CD137 (4-1BB), and their role in the generation of anti-tumor immune responses" Eur J Immunol 32(12): 3617-3627. PubMed
This study addresses the relative importance of CD134 (OX40) and CD137 (4-1BB) in the costimulation of CD4+ and CD8+ T cells under comparable conditions of antigenic stimulation. We demonstrate that CD134 is capable of directly costimulating CD8+ T cells. However, costimulation of CD8+ T cells by CD134 is less potent than that triggered by CD137. The higher costimulatory activity of CD137, when compared with CD134, correlates well with its faster expression kinetics and higher levels on CD8+ T cells. Furthermore, induction of CD137 expression on CD8+ T cells is highly sensitive to low levels of TCR stimulation, which is in contrast with CD134. Conversely, CD134 is more effective than CD137 in costimulating CD4+ T cells. This, however, could not be attributed to differential expression. We also demonstrate that the transient nature of CD134 and CD137 expression on activated CD4+ T cells is the resultof proteolytic shedding. Consistent with the greater ability of CD137 to costimulate CD8+ T cells, stimulation of CD137 in vivo is considerably more effective than CD134 in augmenting anti-tumor immune responses. Therefore, agents that stimulate signaling via CD137 are likely to be more useful in clinical conditions where highly effective CD8+ CTL responses are required.