InVivoPlus anti-mouse PD-L1 (B7-H1)

Product Details
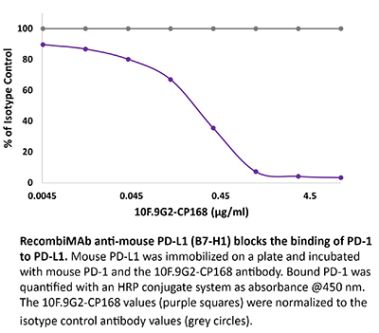
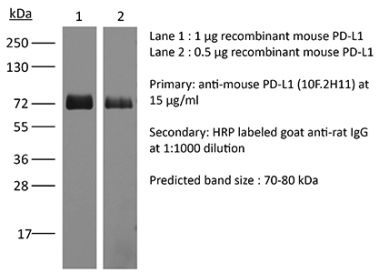
The 10F.9G2™ monoclonal antibody reacts with mouse PD-L1 (programmed death ligand 1) also known as B7-H1 or CD274. PD-L1 is a 40 kDa type I transmembrane protein that belongs to the B7 family of the Ig superfamily. PD-L1 is expressed on T lymphocytes, B lymphocytes, NK cells, dendritic cells, as well as IFNγ stimulated monocytes, epithelial cells and endothelial cells. PD-L1 binds to its receptor, PD-1, found on CD4 and CD8 thymocytes as well as activated T and B lymphocytes and myeloid cells. Engagement of PD-L1 with PD-1 leads to inhibition of TCR-mediated T cell proliferation and cytokine production. PD-L1 is thought to play an important role in tumor immune evasion. Induced PD-L1 expression is common in many tumors and results in increased resistance of tumor cells to CD8 T cell mediated lysis. In mouse models of melanoma, tumor growth can be transiently arrested via treatment with antibodies which block the interaction between PD-L1 and PD-1. The 10F.9G2™ antibody has been shown to block the interaction between PD-L1 and PD-1 and between PD-L1 and B7-1 (CD80).Specifications
| Isotype | Rat IgG2b, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoPlus rat IgG2b isotype control, anti-keyhole limpet hemocyanin |
| Recommended Dilution Buffer | InVivoPure pH 6.5 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Mouse CD274 |
| Reported Applications |
in vivo PD-L1 blockade Immunofluorescence Immunohistochemistry (frozen) Flow cytometry Western blot |
| Formulation |
PBS, pH 6.5 Contains no stabilizers or preservatives |
| Endotoxin* |
<1EU/mg (<0.001EU/μg) Determined by LAL gel clotting assay |
| Aggregation* |
<5% Determined by SEC |
| Purity |
>95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_10949073 |
| Molecular Weight | 150 kDa |
| Murine Pathogen Tests* |
Ectromelia/Mousepox Virus: Negative Hantavirus: Negative K Virus: Negative Lactate Dehydrogenase-Elevating Virus: Negative Lymphocytic Choriomeningitis virus: Negative Mouse Adenovirus: Negative Mouse Cytomegalovirus: Negative Mouse Hepatitis Virus: Negative Mouse Minute Virus: Negative Mouse Norovirus: Negative Mouse Parvovirus: Negative Mouse Rotavirus: Negative Mycoplasma Pulmonis: Negative Pneumonia Virus of Mice: Negative Polyoma Virus: Negative Reovirus Screen: Negative Sendai Virus: Negative Theiler’s Murine Encephalomyelitis: Negative |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
Additional Formats



Recommended Products
-

Recommended Isotype Control(s)
InVivoPlus rat IgG2b isotype control, anti-keyhole limpet hemocyanin
-

Recommended Dilution Buffer
InVivoPure pH 6.5 Dilution Buffer
in vivo PD-L1 blockade
Grasselly, C., et al. (2018). "The Antitumor Activity of Combinations of Cytotoxic Chemotherapy and Immune Checkpoint Inhibitors Is Model-Dependent" Front Immunol 9: 2100. PubMed
In spite of impressive response rates in multiple cancer types, immune checkpoint inhibitors (ICIs) are active in only a minority of patients. Alternative strategies currently aim to combine immunotherapies with conventional agents such as cytotoxic chemotherapies. Here, we performed a study of PD-1 or PDL-1 blockade in combination with reference chemotherapies in four fully immunocompetent mouse models of cancer. We analyzed both the in vivo antitumor response, and the tumor immune infiltrate 4 days after the first treatment. in vivo tumor growth experiments revealed variable responsiveness to ICIs between models. We observed enhanced antitumor effects of the combination of immunotherapy with chemotherapy in the MC38 colon and MB49 bladder models, a lack of response in the 4T1 breast model, and an inhibition of ICIs activity in the MBT-2 bladder model. Flow cytometry analysis of tumor samples showed significant differences in all models between untreated and treated mice. At baseline, all the tumor models studied were predominantly infiltrated with cells harboring an immunosuppressive phenotype. Early alterations of the tumor immune infiltrate after treatment were found to be highly variable. We found that the balance between effector cells and immunosuppressive cells in the tumor microenvironment could be altered with some treatment combinations, but this effect was not always correlated with an impact on in vivo tumor growth. These results show that the combination of cytotoxic chemotherapy with ICIs may result in enhanced, similar or reduced antitumor activity, in a model- and regimen-dependent fashion. The present investigations should help to select appropriate combination regimens for ICIs.
in vivo PD-L1 blockade
Stathopoulou, C., et al. (2018). "PD-1 Inhibitory Receptor Downregulates Asparaginyl Endopeptidase and Maintains Foxp3 Transcription Factor Stability in Induced Regulatory T Cells" Immunity 49(2): 247-263 e247. PubMed
CD4(+) T cell differentiation into multiple T helper (Th) cell lineages is critical for optimal adaptive immune responses. This report identifies an intrinsic mechanism by which programmed death-1 receptor (PD-1) signaling imparted regulatory phenotype to Foxp3(+) Th1 cells (denoted as Tbet(+)iTregPDL1 cells) and inducible regulatory T (iTreg) cells. Tbet(+)iTregPDL1 cells prevented inflammation in murine models of experimental colitis and experimental graft versus host disease (GvHD). Programmed death ligand-1 (PDL-1) binding to PD-1 imparted regulatory function to Tbet(+)iTregPDL1 cells and iTreg cells by specifically downregulating endo-lysosomal protease asparaginyl endopeptidase (AEP). AEP regulated Foxp3 stability and blocking AEP imparted regulatory function in Tbet(+)iTreg cells. Also, Aep(-/-) iTreg cells significantly inhibited GvHD and maintained Foxp3 expression. PD-1-mediated Foxp3 maintenance in Tbet(+) Th1 cells occurred both in tumor infiltrating lymphocytes (TILs) and during chronic viral infection. Collectively, this report has identified an intrinsic function for PD-1 in maintaining Foxp3 through proteolytic pathway.
in vivo PD-L1 blockade, Flow Cytometry
Aloulou, M., et al. (2016). "Follicular regulatory T cells can be specific for the immunizing antigen and derive from naive T cells" Nat Commun 7: 10579. PubMed
T follicular regulatory (Tfr) cells are a subset of Foxp3(+) regulatory T (Treg) cells that form in response to immunization or infection, which localize to the germinal centre where they control the magnitude of the response. Despite an increased interest in the role of Tfr cells in humoral immunity, many fundamental aspects of their biology remain unknown, including whether they recognize self- or foreign antigen. Here we show that Tfr cells can be specific for the immunizing antigen, irrespective of whether it is a self- or foreign antigen. We show that, in addition to developing from thymic derived Treg cells, Tfr cells can also arise from Foxp3(-) precursors in a PD-L1-dependent manner, if the adjuvant used is one that supports T-cell plasticity. These findings have important implications for Tfr cell biology and for improving vaccine efficacy by formulating vaccines that modify the Tfr:Tfh cell ratio.
in vivo PD-L1 blockade, Flow Cytometry
Ngiow, S. F., et al. (2015). "A Threshold Level of Intratumor CD8+ T-cell PD1 Expression Dictates Therapeutic Response to Anti-PD1" Cancer Res 75(18): 3800-3811. PubMed
Despite successes, thus far, a significant proportion of the patients treated with anti-PD1 antibodies have failed to respond. We use mouse tumor models of anti-PD1 sensitivity and resistance and flow cytometry to assess tumor-infiltrating immune cells immediately after therapy. We demonstrate that the expression levels of T-cell PD1 (PD1(lo)), myeloid, and T-cell PDL1 (PDL1(hi)) in the tumor microenvironment inversely correlate and dictate the efficacy of anti-PD1 mAb and function of intratumor CD8(+) T cells. In sensitive tumors, we reveal a threshold for PD1 downregulation on tumor-infiltrating CD8(+) T cells below which the release of adaptive immune resistance is achieved. In contrast, PD1(hi) T cells in resistant tumors fail to be rescued by anti-PD1 therapy and remain dysfunctional unless intratumor PDL1(lo) immune cells are targeted. Intratumor Tregs are partly responsible for the development of anti-PD1-resistant tumors and PD1(hi) CD8(+) T cells. Our analyses provide a framework to interrogate intratumor CD8(+) T-cell PD1 and immune PDL1 levels and response in human cancer. Cancer Res; 75(18); 3800-11. (c)2015 AACR.
in vivo PD-L1 blockade
Jaworska, K., et al. (2015). "Both PD-1 ligands protect the kidney from ischemia reperfusion injury" J Immunol 194(1): 325-333. PubMed
Acute kidney injury (AKI) is a common problem in hospitalized patients that enhances morbidity and mortality and promotes the development of chronic and end-stage renal disease. Ischemia reperfusion injury (IRI) is one of the major causes of AKI and is characterized by uncontrolled renal inflammation and tubular epithelial cell death. Our recent studies demonstrated that regulatory T cells (Tregs) protect the kidney from ischemia reperfusion-induced inflammation and injury. Blockade of programmed death-1 (PD-1) on the surface of Tregs, prior to adoptive transfer, negates their ability to protect against ischemic kidney injury. The present study was designed to investigate the role of the known PD-1 ligands, PD-L1 and PD-L2, in kidney IRI. Administration of PD-L1 or PD-L2 blocking Abs prior to mild or moderate kidney IRI significantly exacerbated the loss of renal function, renal inflammation, and acute tubular necrosis compared with mice receiving isotype control Abs. Interestingly, blockade of both PD-1 ligands resulted in worse injury, dysfunction, and inflammation than did blocking either ligand alone. Genetic deficiency of either PD-1 ligand also exacerbated kidney dysfunction and acute tubular necrosis after subthreshold ischemia. Bone marrow chimeric studies revealed that PD-L1 expressed on non-bone marrow-derived cells is critical for this resistance to IRI. Finally, blockade of either PD-1 ligand negated the protective ability of adoptively transferred Tregs in IRI. These findings suggest that PD-L1 and PD-L2 are nonredundant aspects of the natural protective response to ischemic injury and may be novel therapeutic targets for AKI.
in vivo PD-L1 blockade
Kim, J., et al. (2015). "Memory programming in CD8(+) T-cell differentiation is intrinsic and is not determined by CD4 help" Nat Commun 6: 7994. PubMed
CD8(+) T cells activated without CD4(+) T-cell help are impaired in memory expansion. To understand the underlying cellular mechanism, here we track the dynamics of helper-deficient CD8(+) T-cell response to a minor histocompatibility antigen by phenotypic and in vivo imaging analyses. Helper-deficient CD8(+) T cells show reduced burst expansion, rapid peripheral egress, delayed antigen clearance and continuous activation, and are eventually exhausted. Contrary to the general consensus that CD4 help encodes memory programmes in CD8(+) T cells and helper-deficient CD8(+) T cells are abortive, these cells can differentiate into effectors and memory precursors. Importantly, accelerating antigen clearance or simply increasing the burst effector size enables generation of memory cells by CD8(+) T cells, regardless of CD4 help. These results suggest that the memory programme is CD8(+) T-cell-intrinsic, and provide insight into the role of CD4 help in CD8(+) T-cell responses.
in vivo PD-L1 blockade
Zander, R. A., et al. (2015). "PD-1 Co-inhibitory and OX40 Co-stimulatory Crosstalk Regulates Helper T Cell Differentiation and Anti-Plasmodium Humoral Immunity" Cell Host Microbe 17(5): 628-641. PubMed
The differentiation and protective capacity of Plasmodium-specific T cells are regulated by both positive and negative signals during malaria, but the molecular and cellular details remain poorly defined. Here we show that malaria patients and Plasmodium-infected rodents exhibit atypical expression of the co-stimulatory receptor OX40 on CD4 T cells and that therapeutic enhancement of OX40 signaling enhances helper CD4 T cell activity, humoral immunity, and parasite clearance in rodents. However, these beneficial effects of OX40 signaling are abrogated following coordinate blockade of PD-1 co-inhibitory pathways, which are also upregulated during malaria and associated with elevated parasitemia. Co-administration of biologics blocking PD-1 and promoting OX40 signaling induces excessive interferon-gamma that directly limits helper T cell-mediated support of humoral immunity and decreases parasite control. Our results show that targeting OX40 can enhance Plasmodium control and that crosstalk between co-inhibitory and co-stimulatory pathways in pathogen-specific CD4 T cells can impact pathogen clearance.
in vivo PD-L1 blockade
Tkachev, V., et al. (2015). "Programmed death-1 controls T cell survival by regulating oxidative metabolism" J Immunol 194(12): 5789-5800. PubMed
The coinhibitory receptor programmed death-1 (PD-1) maintains immune homeostasis by negatively regulating T cell function and survival. Blockade of PD-1 increases the severity of graft-versus-host disease (GVHD), but the interplay between PD-1 inhibition and T cell metabolism is not well studied. We found that both murine and human alloreactive T cells concomitantly upregulated PD-1 expression and increased levels of reactive oxygen species (ROS) following allogeneic bone marrow transplantation. This PD-1(Hi)ROS(Hi) phenotype was specific to alloreactive T cells and was not observed in syngeneic T cells during homeostatic proliferation. Blockade of PD-1 signaling decreased both mitochondrial H2O2 and total cellular ROS levels, and PD-1-driven increases in ROS were dependent upon the oxidation of fatty acids, because treatment with etomoxir nullified changes in ROS levels following PD-1 blockade. Downstream of PD-1, elevated ROS levels impaired T cell survival in a process reversed by antioxidants. Furthermore, PD-1-driven changes in ROS were fundamental to establishing a cell’s susceptibility to subsequent metabolic inhibition, because blockade of PD-1 decreased the efficacy of later F1F0-ATP synthase modulation. These data indicate that PD-1 facilitates apoptosis in alloreactive T cells by increasing ROS in a process dependent upon the oxidation of fat. In addition, blockade of PD-1 undermines the potential for subsequent metabolic inhibition, an important consideration given the increasing use of anti-PD-1 therapies in the clinic.
in vivo PD-L1 blockade
Twyman-Saint Victor, C., et al. (2015). "Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer" Nature 520(7547): 373-377. PubMed
Immune checkpoint inhibitors result in impressive clinical responses, but optimal results will require combination with each other and other therapies. This raises fundamental questions about mechanisms of non-redundancy and resistance. Here we report major tumour regressions in a subset of patients with metastatic melanoma treated with an anti-CTLA4 antibody (anti-CTLA4) and radiation, and reproduced this effect in mouse models. Although combined treatment improved responses in irradiated and unirradiated tumours, resistance was common. Unbiased analyses of mice revealed that resistance was due to upregulation of PD-L1 on melanoma cells and associated with T-cell exhaustion. Accordingly, optimal response in melanoma and other cancer types requires radiation, anti-CTLA4 and anti-PD-L1/PD-1. Anti-CTLA4 predominantly inhibits T-regulatory cells (Treg cells), thereby increasing the CD8 T-cell to Treg (CD8/Treg) ratio. Radiation enhances the diversity of the T-cell receptor (TCR) repertoire of intratumoral T cells. Together, anti-CTLA4 promotes expansion of T cells, while radiation shapes the TCR repertoire of the expanded peripheral clones. Addition of PD-L1 blockade reverses T-cell exhaustion to mitigate depression in the CD8/Treg ratio and further encourages oligoclonal T-cell expansion. Similarly to results from mice, patients on our clinical trial with melanoma showing high PD-L1 did not respond to radiation plus anti-CTLA4, demonstrated persistent T-cell exhaustion, and rapidly progressed. Thus, PD-L1 on melanoma cells allows tumours to escape anti-CTLA4-based therapy, and the combination of radiation, anti-CTLA4 and anti-PD-L1 promotes response and immunity through distinct mechanisms.
in vivo PD-L1 blockade, Flow Cytometry
Rutigliano, J. A., et al. (2014). "Highly pathological influenza A virus infection is associated with augmented expression of PD-1 by functionally compromised virus-specific CD8+ T cells" J Virol 88(3): 1636-1651. PubMed
One question that continues to challenge influenza A research is why some strains of virus are so devastating compared to their more mild counterparts. We approached this question from an immunological perspective, investigating the CD8(+) T cell response in a mouse model system comparing high- and low-pathological influenza virus infections. Our findings reveal that the early (day 0 to 5) viral titer was not the determining factor in the outcome of disease. Instead, increased numbers of antigen-specific CD8(+) T cells and elevated effector function on a per-cell basis were found in the low-pathological infection and correlated with reduced illness and later-time-point (day 6 to 10) viral titer. High-pathological infection was associated with increased PD-1 expression on influenza virus-specific CD8(+) T cells, and blockade of PD-L1 in vivo led to reduced virus titers and increased CD8(+) T cell numbers in high- but not low-pathological infection, though T cell functionality was not restored. These data show that high-pathological acute influenza virus infection is associated with a dysregulated CD8(+) T cell response, which is likely caused by the more highly inflamed airway microenvironment during the early days of infection. Therapeutic approaches specifically aimed at modulating innate airway inflammation may therefore promote efficient CD8(+) T cell activity. We show that during a severe influenza virus infection, one type of immune cell, the CD8 T cell, is less abundant and less functional than in a more mild infection. This dysregulated T cell phenotype correlates with a lower rate of virus clearance in the severe infection and is partially regulated by the expression of a suppressive coreceptor called PD-1. Treatment with an antibody that blocks PD-1 improves T cell functionality and increases virus clearance.
in vivo PD-L1 blockade
Yang, X., et al. (2014). "Targeting the tumor microenvironment with interferon-beta bridges innate and adaptive immune responses" Cancer Cell 25(1): 37-48. PubMed
Antibodies (Abs) that preferentially target oncogenic receptors have been increasingly used for cancer therapy, but tumors often acquire intrinsic Ab resistance after prolonged and costly treatment. Herein we armed the Ab with IFNbeta and observed that it is more potent than the first generation of Ab for controlling Ab-resistant tumors. This strategy controls Ab resistance by rebridging suppressed innate and adaptive immunity in the tumor microenvironment. Mechanistically, Ab-IFNbeta therapy primarily and directly targets intratumoral dendritic cells, which reactivate CTL by increasing antigen cross-presentation within the tumor microenvironment. Additionally, blocking PD-L1, which is induced by Ab-IFNbeta treatment, overcomes treatment-acquired resistance and completely eradicates established tumors. This study establishes a next-generation Ab-based immunotherapy that targets and eradicates established Ab-resistant tumors.
in vivo PD-L1 blockade
Dolina, J. S., et al. (2014). "Liver-primed CD8+ T cells suppress antiviral adaptive immunity through galectin-9-independent T-cell immunoglobulin and mucin 3 engagement of high-mobility group box 1 in mice" Hepatology 59(4): 1351-1365. PubMed
The liver is a tolerogenic environment exploited by persistent infections, such as hepatitis B (HBV) and C (HCV) viruses. In a murine model of intravenous hepatotropic adenovirus infection, liver-primed antiviral CD8(+) T cells fail to produce proinflammatory cytokines and do not display cytolytic activity characteristic of effector CD8(+) T cells generated by infection at an extrahepatic, that is, subcutaneous, site. Importantly, liver-generated CD8(+) T cells also appear to have a T-regulatory (Treg) cell function exemplified by their ability to limit proliferation of antigen-specific T-effector (Teff ) cells in vitro and in vivo via T-cell immunoglobulin and mucin 3 (Tim-3) expressed by the CD8(+) Treg cells. Regulatory activity did not require recognition of the canonical Tim-3 ligand, galectin-9, but was dependent on CD8(+) Treg cell-surface Tim-3 binding to the alarmin, high-mobility group box 1 (HMGB-1). CONCLUSION: Virus-specific Tim-3(+) CD8(+) T cells operating through HMGB-1 recognition in the setting of acute and chronic viral infections of the liver may act to dampen hepatic T-cell responses in the liver microenvironment and, as a consequence, limit immune-mediated tissue injury or promote the establishment of persistent infections.
in vivo PD-L1 blockade
Deng, L., et al. (2014). "Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice" J Clin Invest 124(2): 687-695. PubMed
High-dose ionizing irradiation (IR) results in direct tumor cell death and augments tumor-specific immunity, which enhances tumor control both locally and distantly. Unfortunately, local relapses often occur following IR treatment, indicating that IR-induced responses are inadequate to maintain antitumor immunity. Therapeutic blockade of the T cell negative regulator programmed death-ligand 1 (PD-L1, also called B7-H1) can enhance T cell effector function when PD-L1 is expressed in chronically inflamed tissues and tumors. Here, we demonstrate that PD-L1 was upregulated in the tumor microenvironment after IR. Administration of anti-PD-L1 enhanced the efficacy of IR through a cytotoxic T cell-dependent mechanism. Concomitant with IR-mediated tumor regression, we observed that IR and anti-PD-L1 synergistically reduced the local accumulation of tumor-infiltrating myeloid-derived suppressor cells (MDSCs), which suppress T cells and alter the tumor immune microenvironment. Furthermore, activation of cytotoxic T cells with combination therapy mediated the reduction of MDSCs in tumors through the cytotoxic actions of TNF. Our data provide evidence for a close interaction between IR, T cells, and the PD-L1/PD-1 axis and establish a basis for the rational design of combination therapy with immune modulators and radiotherapy.
in vivo PD-L1 blockade
Dietze, K. K., et al. (2013). "Combining regulatory T cell depletion and inhibitory receptor blockade improves reactivation of exhausted virus-specific CD8+ T cells and efficiently reduces chronic retroviral loads" PLoS Pathog 9(12): e1003798. PubMed
Chronic infections with human viruses, such as HIV and HCV, or mouse viruses, such as LCMV or Friend Virus (FV), result in functional exhaustion of CD8(+) T cells. Two main mechanisms have been described that mediate this exhaustion: expression of inhibitory receptors on CD8(+) T cells and expansion of regulatory T cells (Tregs) that suppress CD8(+) T cell activity. Several studies show that blockage of one of these pathways results in reactivation of CD8(+) T cells and partial reduction in chronic viral loads. Using blocking antibodies against PD-1 ligand and Tim-3 and transgenic mice in which Tregs can be selectively ablated, we compared these two treatment strategies and combined them for the first time in a model of chronic retrovirus infection. Blocking inhibitory receptors was more efficient than transient depletion of Tregs in reactivating exhausted CD8(+) T cells and reducing viral set points. However, a combination therapy was superior to any single treatment and further augmented CD8(+) T cell responses and resulted in a sustained reduction in chronic viral loads. These results demonstrate that Tregs and inhibitory receptors are non-overlapping factors in the maintenance of chronic viral infections and that immunotherapies targeting both pathways may be a promising strategy to treat chronic infectious diseases.
in vivo PD-L1 blockade, Immunofluorescence
Willimsky, G., et al. (2013). "Virus-induced hepatocellular carcinomas cause antigen-specific local tolerance" J Clin Invest 123(3): 1032-1043. PubMed
T cell surveillance is often effective against virus-associated tumors because of their high immunogenicity. It is not clear why surveillance occasionally fails, particularly against hepatitis B virus- or hepatitis C virus-associated hepatocellular carcinoma (HCC). We established a transgenic murine model of virus-induced HCC by hepatocyte-specific adenovirus-induced activation of the oncogenic SV40 large T antigen (TAg). Adenovirus infection induced cytotoxic T lymphocytes (CTLs) targeted against the virus and TAg, leading to clearance of the infected cells. Despite the presence of functional, antigen-specific T cells, a few virus-infected cells escaped immune clearance and progressed to HCC. These cells expressed TAg at levels similar to HCC isolated from neonatal TAg-tolerant mice, suggesting that CTL clearance does not select for cells with low immunogenicity. Virus-infected mice revealed significantly greater T cell infiltration in early-stage HCC compared with that in late-stage HCC, demonstrating progressive local immune suppression through inefficient T cell infiltration. Programmed cell death protein-1 (PD-1) and its ligand PD-L1 were expressed in all TAg-specific CD8+ T cells and HCC, respectively, which contributed to local tumor-antigen-specific tolerance. Thus, we have developed a model of virus-induced HCC that may allow for a better understanding of human HCC.
in vivo PD-L1 blockade
Hafalla, J. C., et al. (2012). "The CTLA-4 and PD-1/PD-L1 inhibitory pathways independently regulate host resistance to Plasmodium-induced acute immune pathology" PLoS Pathog 8(2): e1002504. PubMed
The balance between pro-inflammatory and regulatory immune responses in determining optimal T cell activation is vital for the successful resolution of microbial infections. This balance is maintained in part by the negative regulators of T cell activation, CTLA-4 and PD-1/PD-L, which dampen effector responses during chronic infections. However, their role in acute infections, such as malaria, remains less clear. In this study, we determined the contribution of CTLA-4 and PD-1/PD-L to the regulation of T cell responses during Plasmodium berghei ANKA (PbA)-induced experimental cerebral malaria (ECM) in susceptible (C57BL/6) and resistant (BALB/c) mice. We found that the expression of CTLA-4 and PD-1 on T cells correlates with the extent of pro-inflammatory responses induced during PbA infection, being higher in C57BL/6 than in BALB/c mice. Thus, ECM develops despite high levels of expression of these inhibitory receptors. However, antibody-mediated blockade of either the CTLA-4 or PD-1/PD-L1, but not the PD-1/PD-L2, pathways during PbA-infection in ECM-resistant BALB/c mice resulted in higher levels of T cell activation, enhanced IFN-gamma production, increased intravascular arrest of both parasitised erythrocytes and CD8(+) T cells to the brain, and augmented incidence of ECM. Thus, in ECM-resistant BALB/c mice, CTLA-4 and PD-1/PD-L1 represent essential, independent and non-redundant pathways for maintaining T cell homeostasis during a virulent malaria infection. Moreover, neutralisation of IFN-gamma or depletion of CD8(+) T cells during PbA infection was shown to reverse the pathologic effects of regulatory pathway blockade, highlighting that the aetiology of ECM in the BALB/c mice is similar to that in C57BL/6 mice. In summary, our results underscore the differential and complex regulation that governs immune responses to malaria parasites.
Immunohistochemistry (frozen), Immunofluorescence
Riella, L. V., et al. (2011). "Essential role of PDL1 expression on nonhematopoietic donor cells in acquired tolerance to vascularized cardiac allografts" Am J Transplant 11(4): 832-840. PubMed
The PD1:PDL1 pathway is an essential negative costimulatory pathway that plays a key role in regulating the alloimune response. PDL1 is expressed not only on antigen-presenting cells (APCs) but also cardiac endothelium. In this study, we investigated the importance of PDL1 expression on donor cardiac allograft in acquired transplantation tolerance in a fully MHC-mismatched model. We generated PDL1 chimeric mice on B6 background that expressed PDL1 on either hematopoietic cells or nonhematopoietic cells of the heart. Sham animals were used as controls. These hearts were then transplanted into BALB/c recipients and treated with CTLA4-Ig to induce tolerance. Cardiac endothelium showed significant expression of PDL1, which was upregulated upon transplantation. While the absence of PDL1 on hematopoietic cells of the heart resulted in delayed rejection and prevented long-term tolerance in most but not all recipients, we observed an accelerated and early graft rejection of all donor allografts that lacked PDL1 on the endothelium. Moreover, PDL1-deficient endothelium hearts had significant higher frequency of IFN-gamma-producing alloreactive cells as well as higher frequency of CD8(+) effector T cells. These findings demonstrate that PDL1 expression mainly on donor endothelium is functionally important in a fully allogeneic mismatched model for the induction of cardiac allograft tolerance
in vivo PD-L1 blockade
Zhang, L., et al. (2009). "PD-1/PD-L1 interactions inhibit antitumor immune responses in a murine acute myeloid leukemia model" Blood 114(8): 1545-1552. PubMed
Negative regulatory mechanisms within the solid tumor microenvironment inhibit antitumor T-cell function, leading to evasion from immune attack. One inhibitory mechanism is up-regulation of programmed death-ligand 1 (PD-L1) expressed on tumor or stromal cells which binds to programmed death-1 (PD-1) on activated T cells. PD-1/PD-L1 engagement results in diminished antitumor T-cell responses and correlates with poor outcome in murine and human solid cancers. In contrast to available data in solid tumors, little is known regarding involvement of the PD-1/PD-L1 pathway in immune escape by hematopoietic cancers, such as acute myeloid leukemia (AML). To investigate this hypothesis, we used the murine leukemia, C1498. When transferred intravenously, C1498 cells grew progressively and apparently evaded immune destruction. Low levels of PD-L1 expression were found on C1498 cells grown in vitro. However, PD-L1 expression was up-regulated on C1498 cells when grown in vivo. PD-1(-/-) mice challenged with C1498 cells generated augmented antitumor T-cell responses, showed decreased AML burden in the blood and other organs, and survived significantly longer than did wild-type mice. Similar results were obtained with a PD-L1 blocking antibody. These data suggest the importance of the PD-1/PD-L1 pathway in immune evasion by a hematologic malignancy, providing a rationale for clinical trials targeting this pathway in leukemia patients.