InVivoPlus anti-mouse CD8α

Product Details
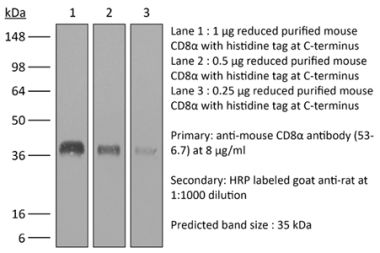
The 2.43 monoclonal antibody reacts with mouse CD8α. The CD8 antigen is a transmembrane glycoprotein that acts as a co-receptor for the T cell receptor (TCR). Like the TCR, CD8 binds to class I MHC molecules displayed by antigen presenting cells (APC). CD8 is primarily expressed on the surface of cytotoxic T cells, but can also be found on thymocytes, natural killer cells, and some dendritic cell subsets. CD8 most commonly exists as a heterodimer composed of one CD8α and one CD8β chain however, it can also exist as a homodimer composed of two CD8α chains. Both the CD8α and CD8β chains share significant homology to immunoglobulin variable light chains. The molecular weight of each CD8 chain is approximately 34 kDa. The 2.43 antibody exhibits depleting activity when used in vivo.Specifications
| Isotype | Rat IgG2b, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoPlus rat IgG2b isotype control, anti-keyhole limpet hemocyanin |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Mouse CTL clone L3 |
| Reported Applications |
in vivo CD8+ T cell depletion Western blot |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin* |
<1EU/mg (<0.001EU/μg) Determined by LAL gel clotting assay |
| Aggregation* |
<5% Determined by SEC |
| Purity |
>95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_1125541 |
| Molecular Weight | 150 kDa |
| Murine Pathogen Tests* |
Ectromelia/Mousepox Virus: Negative Hantavirus: Negative K Virus: Negative Lactate Dehydrogenase-Elevating Virus: Negative Lymphocytic Choriomeningitis virus: Negative Mouse Adenovirus: Negative Mouse Cytomegalovirus: Negative Mouse Hepatitis Virus: Negative Mouse Minute Virus: Negative Mouse Norovirus: Negative Mouse Parvovirus: Negative Mouse Rotavirus: Negative Mycoplasma Pulmonis: Negative Pneumonia Virus of Mice: Negative Polyoma Virus: Negative Reovirus Screen: Negative Sendai Virus: Negative Theiler’s Murine Encephalomyelitis: Negative |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
Additional Formats


Recommended Products
-

Recommended Isotype Control(s)
InVivoPlus rat IgG2b isotype control, anti-keyhole limpet hemocyanin
-

Recommended Dilution Buffer
InVivoPure pH 7.0 Dilution Buffer
in vivo CD8+ T cell depletion
Balogh, K. N., et al. (2018). "Macrophage Migration Inhibitory Factor protects cancer cells from immunogenic cell death and impairs anti-tumor immune responses" PLoS One 13(6): e0197702. PubMed
The Macrophage Migration Inhibitory Factor (MIF) is an inflammatory cytokine that is overexpressed in a number of cancer types, with increased MIF expression often correlating with tumor aggressiveness and poor patient outcomes. In this study, we aimed to better understand the link between primary tumor expression of MIF and increased tumor growth. Using the MMTV-PyMT murine model of breast cancer, we observed that elevated MIF expression promoted tumor appearance and growth. Supporting this, we confirmed our previous observation that higher MIF expression supported tumor growth in the 4T1 murine model of breast cancer. We subsequently discovered that loss of MIF expression in 4T1 cells led to decreased cell numbers and increased apoptosis in vitro under reduced serum culture conditions. We hypothesized that this increase in cell death would promote detection by the host immune system in vivo, which could explain the observed impairment in tumor growth. Supporting this, we demonstrated that loss of MIF expression in the primary tumor led to an increased abundance of intra-tumoral IFNgamma-producing CD4+ and CD8+ T cells, and that depletion of T cells from mice bearing MIF-deficient tumors restored growth to the level of MIF-expressing tumors. Furthermore, we found that MIF depletion from the tumor cells resulted in greater numbers of activated intra-tumoral dendritic cells (DCs). Lastly, we demonstrated that loss of MIF expression led to a robust induction of a specialized form of cell death, immunogenic cell death (ICD), in vitro. Together, our data suggests a model in which MIF expression in the primary tumor dampens the anti-tumor immune response, promoting tumor growth.
in vivo CD8+ T cell depletion
Li, J., et al. (2018). "Co-inhibitory Molecule B7 Superfamily Member 1 Expressed by Tumor-Infiltrating Myeloid Cells Induces Dysfunction of Anti-tumor CD8(+) T Cells" Immunity 48(4): 773-786 e775. PubMed
The molecular mechanisms whereby CD8(+) T cells become “exhausted” in the tumor microenvironment remain unclear. Programmed death ligand-1 (PD-L1) is upregulated on tumor cells and PD-1-PD-L1 blockade has significant efficacy in human tumors; however, most patients do not respond, suggesting additional mechanisms underlying T cell exhaustion. B7 superfamily member 1 (B7S1), also called B7-H4, B7x, or VTCN1, negatively regulates T cell activation. Here we show increased B7S1 expression on myeloid cells from human hepatocellular carcinoma correlated with CD8(+) T cell dysfunction. B7S1 inhibition suppressed development of murine tumors. Putative B7S1 receptor was co-expressed with PD-1 but not T cell immunoglobulin and mucin-domain containing-3 (Tim-3) at an activated state of early tumor-infiltrating CD8(+) T cells, and B7S1 promoted T cell exhaustion, possibly through Eomes overexpression. Combinatorial blockade of B7S1 and PD-1 synergistically enhanced anti-tumor immune responses. Collectively, B7S1 initiates dysfunction of tumor-infiltrating CD8(+) T cells and may be targeted for cancer immunotherapy.
in vivo CD8+ T cell depletion
Moynihan, K. D., et al. (2016). "Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses" Nat Med. doi : 10.1038/nm.4200. PubMed
Checkpoint blockade with antibodies specific for cytotoxic T lymphocyte-associated protein (CTLA)-4 or programmed cell death 1 (PDCD1; also known as PD-1) elicits durable tumor regression in metastatic cancer, but these dramatic responses are confined to a minority of patients. This suboptimal outcome is probably due in part to the complex network of immunosuppressive pathways present in advanced tumors, which are unlikely to be overcome by intervention at a single signaling checkpoint. Here we describe a combination immunotherapy that recruits a variety of innate and adaptive immune cells to eliminate large tumor burdens in syngeneic tumor models and a genetically engineered mouse model of melanoma; to our knowledge tumors of this size have not previously been curable by treatments relying on endogenous immunity. Maximal antitumor efficacy required four components: a tumor-antigen-targeting antibody, a recombinant interleukin-2 with an extended half-life, anti-PD-1 and a powerful T cell vaccine. Depletion experiments revealed that CD8+ T cells, cross-presenting dendritic cells and several other innate immune cell subsets were required for tumor regression. Effective treatment induced infiltration of immune cells and production of inflammatory cytokines in the tumor, enhanced antibody-mediated tumor antigen uptake and promoted antigen spreading. These results demonstrate the capacity of an elicited endogenous immune response to destroy large, established tumors and elucidate essential characteristics of combination immunotherapies that are capable of curing a majority of tumors in experimental settings typically viewed as intractable.
in vivo CD8+ T cell depletion
Voron, T., et al. (2015). "VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors" J Exp Med 212(2): 139-148. PubMed
Immune escape is a prerequisite for tumor development. To avoid the immune system, tumors develop different mechanisms, including T cell exhaustion, which is characterized by expression of immune inhibitory receptors, such as PD-1, CTLA-4, Tim-3, and a progressive loss of function. The recent development of therapies targeting PD-1 and CTLA-4 have raised great interest since they induced long-lasting objective responses in patients suffering from advanced metastatic tumors. However, the regulation of PD-1 expression, and thereby of exhaustion, is unclear. VEGF-A, a proangiogenic molecule produced by the tumors, plays a key role in the development of an immunosuppressive microenvironment. We report in the present work that VEGF-A produced in the tumor microenvironment enhances expression of PD-1 and other inhibitory checkpoints involved in CD8(+) T cell exhaustion, which could be reverted by anti-angiogenic agents targeting VEGF-A-VEGFR. In view of these results, association of anti-angiogenic molecules with immunomodulators of inhibitory checkpoints may be of particular interest in VEGF-A-producing tumors.
in vivo CD8+ T cell depletion
Vanpouille-Box, C., et al. (2015). "TGFbeta Is a Master Regulator of Radiation Therapy-Induced Antitumor Immunity" Cancer Res 75(11): 2232-2242. PubMed
T cells directed to endogenous tumor antigens are powerful mediators of tumor regression. Recent immunotherapy advances have identified effective interventions to unleash tumor-specific T-cell activity in patients who naturally develop them. Eliciting T-cell responses to a patient’s individual tumor remains a major challenge. Radiation therapy can induce immune responses to model antigens expressed by tumors, but it remains unclear whether it can effectively prime T cells specific for endogenous antigens expressed by poorly immunogenic tumors. We hypothesized that TGFbeta activity is a major obstacle hindering the ability of radiation to generate an in situ tumor vaccine. Here, we show that antibody-mediated TGFbeta neutralization during radiation therapy effectively generates CD8(+) T-cell responses to multiple endogenous tumor antigens in poorly immunogenic mouse carcinomas. Generated T cells were effective at causing regression of irradiated tumors and nonirradiated lung metastases or synchronous tumors (abscopal effect). Gene signatures associated with IFNgamma and immune-mediated rejection were detected in tumors treated with radiation therapy and TGFbeta blockade in combination but not as single agents. Upregulation of programmed death (PD) ligand-1 and -2 in neoplastic and myeloid cells and PD-1 on intratumoral T cells limited tumor rejection, resulting in rapid recurrence. Addition of anti-PD-1 antibodies extended survival achieved with radiation and TGFbeta blockade. Thus, TGFbeta is a fundamental regulator of radiation therapy’s ability to generate an in situ tumor vaccine. The combination of local radiation therapy with TGFbeta neutralization offers a novel individualized strategy for vaccinating patients against their tumors.
in vivo CD8+ T cell depletion
Yamada, D. H., et al. (2015). "Suppression of Fcgamma-receptor-mediated antibody effector function during persistent viral infection" Immunity 42(2): 379-390. PubMed
Understanding how viruses subvert host immunity and persist is essential for developing strategies to eliminate infection. T cell exhaustion during chronic viral infection is well described, but effects on antibody-mediated effector activity are unclear. Herein, we show that increased amounts of immune complexes generated in mice persistently infected with lymphocytic choriomeningitis virus (LCMV) suppressed multiple Fcgamma-receptor (FcgammaR) functions. The high amounts of immune complexes suppressed antibody-mediated cell depletion, therapeutic antibody-killing of LCMV infected cells and human CD20-expressing tumors, as well as reduced immune complex-mediated cross-presentation to T cells. Suppression of FcgammaR activity was not due to inhibitory FcgammaRs or high concentrations of free antibody, and proper FcgammaR functions were restored when persistently infected mice specifically lacked immune complexes. Thus, we identify a mechanism of immunosuppression during viral persistence with implications for understanding effective antibody activity aimed at pathogen control.
in vivo CD8+ T cell depletion
Twyman-Saint Victor, C., et al. (2015). "Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer" Nature 520(7547): 373-377. PubMed
Immune checkpoint inhibitors result in impressive clinical responses, but optimal results will require combination with each other and other therapies. This raises fundamental questions about mechanisms of non-redundancy and resistance. Here we report major tumour regressions in a subset of patients with metastatic melanoma treated with an anti-CTLA4 antibody (anti-CTLA4) and radiation, and reproduced this effect in mouse models. Although combined treatment improved responses in irradiated and unirradiated tumours, resistance was common. Unbiased analyses of mice revealed that resistance was due to upregulation of PD-L1 on melanoma cells and associated with T-cell exhaustion. Accordingly, optimal response in melanoma and other cancer types requires radiation, anti-CTLA4 and anti-PD-L1/PD-1. Anti-CTLA4 predominantly inhibits T-regulatory cells (Treg cells), thereby increasing the CD8 T-cell to Treg (CD8/Treg) ratio. Radiation enhances the diversity of the T-cell receptor (TCR) repertoire of intratumoral T cells. Together, anti-CTLA4 promotes expansion of T cells, while radiation shapes the TCR repertoire of the expanded peripheral clones. Addition of PD-L1 blockade reverses T-cell exhaustion to mitigate depression in the CD8/Treg ratio and further encourages oligoclonal T-cell expansion. Similarly to results from mice, patients on our clinical trial with melanoma showing high PD-L1 did not respond to radiation plus anti-CTLA4, demonstrated persistent T-cell exhaustion, and rapidly progressed. Thus, PD-L1 on melanoma cells allows tumours to escape anti-CTLA4-based therapy, and the combination of radiation, anti-CTLA4 and anti-PD-L1 promotes response and immunity through distinct mechanisms.
in vivo CD8+ T cell depletion
Coffelt, S. B., et al. (2015). "IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis" Nature 522(7556): 345-348. PubMed
Metastatic disease remains the primary cause of death for patients with breast cancer. The different steps of the metastatic cascade rely on reciprocal interactions between cancer cells and their microenvironment. Within this local microenvironment and in distant organs, immune cells and their mediators are known to facilitate metastasis formation. However, the precise contribution of tumour-induced systemic inflammation to metastasis and the mechanisms regulating systemic inflammation are poorly understood. Here we show that tumours maximize their chance of metastasizing by evoking a systemic inflammatory cascade in mouse models of spontaneous breast cancer metastasis. We mechanistically demonstrate that interleukin (IL)-1beta elicits IL-17 expression from gamma delta (gammadelta) T cells, resulting in systemic, granulocyte colony-stimulating factor (G-CSF)-dependent expansion and polarization of neutrophils in mice bearing mammary tumours. Tumour-induced neutrophils acquire the ability to suppress cytotoxic T lymphocytes carrying the CD8 antigen, which limit the establishment of metastases. Neutralization of IL-17 or G-CSF and absence of gammadelta T cells prevents neutrophil accumulation and downregulates the T-cell-suppressive phenotype of neutrophils. Moreover, the absence of gammadelta T cells or neutrophils profoundly reduces pulmonary and lymph node metastases without influencing primary tumour progression. Our data indicate that targeting this novel cancer-cell-initiated domino effect within the immune system–the gammadelta T cell/IL-17/neutrophil axis–represents a new strategy to inhibit metastatic disease.
in vivo CD8+ T cell depletion
Evans, E. E., et al. (2015). "Antibody Blockade of Semaphorin 4D Promotes Immune Infiltration into Tumor and Enhances Response to Other Immunomodulatory Therapies" Cancer Immunol Res 3(6): 689-701. PubMed
Semaphorin 4D (SEMA4D, CD100) and its receptor plexin-B1 (PLXNB1) are broadly expressed in murine and human tumors, and their expression has been shown to correlate with invasive disease in several human tumors. SEMA4D normally functions to regulate the motility and differentiation of multiple cell types, including those of the immune, vascular, and nervous systems. In the setting of cancer, SEMA4D-PLXNB1 interactions have been reported to affect vascular stabilization and transactivation of ERBB2, but effects on immune-cell trafficking in the tumor microenvironment (TME) have not been investigated. We describe a novel immunomodulatory function of SEMA4D, whereby strong expression of SEMA4D at the invasive margins of actively growing tumors influences the infiltration and distribution of leukocytes in the TME. Antibody neutralization of SEMA4D disrupts this gradient of expression, enhances recruitment of activated monocytes and lymphocytes into the tumor, and shifts the balance of cells and cytokines toward a proinflammatory and antitumor milieu within the TME. This orchestrated change in the tumor architecture was associated with durable tumor rejection in murine Colon26 and ERBB2(+) mammary carcinoma models. The immunomodulatory activity of anti-SEMA4D antibody can be enhanced by combination with other immunotherapies, including immune checkpoint inhibition and chemotherapy. Strikingly, the combination of anti-SEMA4D antibody with antibody to CTLA-4 acts synergistically to promote complete tumor rejection and survival. Inhibition of SEMA4D represents a novel mechanism and therapeutic strategy to promote functional immune infiltration into the TME and inhibit tumor progression.
in vivo CD8+ T cell depletion
Van der Jeught, K., et al. (2014). "Intratumoral administration of mRNA encoding a fusokine consisting of IFN-beta and the ectodomain of the TGF-beta receptor II potentiates antitumor immunity" Oncotarget 5(20): 10100-10113. PubMed
It is generally accepted that the success of immunotherapy depends on the presence of tumor-specific CD8(+) cytotoxic T cells and the modulation of the tumor environment. In this study, we validated mRNA encoding soluble factors as a tool to modulate the tumor microenvironment to potentiate infiltration of tumor-specific T cells. Intratumoral delivery of mRNA encoding a fusion protein consisting of interferon-beta and the ectodomain of the transforming growth factor-beta receptor II, referred to as Fbeta(2), showed therapeutic potential. The treatment efficacy was dependent on CD8(+) T cells and could be improved through blockade of PD-1/PD-L1 interactions. In vitro studies revealed that administration of Fbeta(2) to tumor cells resulted in a reduced proliferation and increased expression of MHC I but also PD-L1. Importantly, Fbeta(2) enhanced the antigen presenting capacity of dendritic cells, whilst reducing the suppressive activity of myeloid-derived suppressor cells. In conclusion, these data suggest that intratumoral delivery of mRNA encoding soluble proteins, such as Fbeta(2), can modulate the tumor microenvironment, leading to effective antitumor T cell responses, which can be further potentiated through combination therapy.
in vivo CD8+ T cell depletion
Deng, L., et al. (2014). "Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice" J Clin Invest 124(2): 687-695. PubMed
High-dose ionizing irradiation (IR) results in direct tumor cell death and augments tumor-specific immunity, which enhances tumor control both locally and distantly. Unfortunately, local relapses often occur following IR treatment, indicating that IR-induced responses are inadequate to maintain antitumor immunity. Therapeutic blockade of the T cell negative regulator programmed death-ligand 1 (PD-L1, also called B7-H1) can enhance T cell effector function when PD-L1 is expressed in chronically inflamed tissues and tumors. Here, we demonstrate that PD-L1 was upregulated in the tumor microenvironment after IR. Administration of anti-PD-L1 enhanced the efficacy of IR through a cytotoxic T cell-dependent mechanism. Concomitant with IR-mediated tumor regression, we observed that IR and anti-PD-L1 synergistically reduced the local accumulation of tumor-infiltrating myeloid-derived suppressor cells (MDSCs), which suppress T cells and alter the tumor immune microenvironment. Furthermore, activation of cytotoxic T cells with combination therapy mediated the reduction of MDSCs in tumors through the cytotoxic actions of TNF. Our data provide evidence for a close interaction between IR, T cells, and the PD-L1/PD-1 axis and establish a basis for the rational design of combination therapy with immune modulators and radiotherapy.
in vivo CD8+ T cell depletion
DeBerge, M. P., et al. (2014). "Soluble, but not transmembrane, TNF-alpha is required during influenza infection to limit the magnitude of immune responses and the extent of immunopathology" J Immunol 192(12): 5839-5851. PubMed
TNF-alpha is a pleotropic cytokine that has both proinflammatory and anti-inflammatory functions during influenza infection. TNF-alpha is first expressed as a transmembrane protein that is proteolytically processed to release a soluble form. Transmembrane TNF-alpha (memTNF-alpha) and soluble TNF-alpha (solTNF-alpha) have been shown to exert distinct tissue-protective or tissue-pathologic effects in several disease models. However, the relative contributions of memTNF-alpha or solTNF-alpha in regulating pulmonary immunopathology following influenza infection are unclear. Therefore, we performed intranasal influenza infection in mice exclusively expressing noncleavable memTNF-alpha or lacking TNF-alpha entirely and examined the outcomes. We found that solTNF-alpha, but not memTNF-alpha, was required to limit the size of the immune response and the extent of injury. In the absence of solTNF-alpha, there was a significant increase in the CD8(+) T cell response, including virus-specific CD8(+) T cells, which was due in part to an increased resistance to activation-induced cell death. We found that solTNF-alpha mediates these immunoregulatory effects primarily through TNFR1, because mice deficient in TNFR1, but not TNFR2, exhibited dysregulated immune responses and exacerbated injury similar to that observed in mice lacking solTNF-alpha. We also found that solTNF-alpha expression was required early during infection to regulate the magnitude of the CD8(+) T cell response, indicating that early inflammatory events are critical for the regulation of the effector phase. Taken together, these findings suggest that processing of memTNF-alpha to release solTNF-alpha is a critical event regulating the immune response during influenza infection.
in vivo CD8+ T cell depletion
Vegran, F., et al. (2014). "The transcription factor IRF1 dictates the IL-21-dependent anticancer functions of TH9 cells" Nat Immunol 15(8): 758-766. PubMed
The TH9 subset of helper T cells was initially shown to contribute to the induction of autoimmune and allergic diseases, but subsequent evidence has suggested that these cells also exert antitumor activities. However, the molecular events that account for their effector properties are elusive. Here we found that the transcription factor IRF1 enhanced the effector function of TH9 cells and dictated their anticancer properties. Under TH9-skewing conditions, interleukin 1beta (IL-1beta) induced phosphorylation of the transcription factor STAT1 and subsequent expression of IRF1, which bound to the promoters of Il9 and Il21 and enhanced secretion of the cytokines IL-9 and IL-21 from TH9 cells. Furthermore, IL-1beta-induced TH9 cells exerted potent anticancer functions in an IRF1- and IL-21-dependent manner. Our findings thus identify IRF1 as a target for controlling the function of TH9 cells.
in vivo CD8+ T cell depletion
Sandoval, F., et al. (2013). "Mucosal imprinting of vaccine-induced CD8(+) T cells is crucial to inhibit the growth of mucosal tumors" Sci Transl Med 5(172): 172ra120. PubMed
Although many human cancers are located in mucosal sites, most cancer vaccines are tested against subcutaneous tumors in preclinical models. We therefore wondered whether mucosa-specific homing instructions to the immune system might influence mucosal tumor outgrowth. We showed that the growth of orthotopic head and neck or lung cancers was inhibited when a cancer vaccine was delivered by the intranasal mucosal route but not the intramuscular route. This antitumor effect was dependent on CD8(+) T cells. Indeed, only intranasal vaccination elicited mucosal-specific CD8(+) T cells expressing the mucosal integrin CD49a. Blockade of CD49a decreased intratumoral CD8(+) T cell infiltration and the efficacy of cancer vaccine on mucosal tumor. We then showed that after intranasal vaccination, dendritic cells from lung parenchyma, but not those from spleen, induced the expression of CD49a on cocultured specific CD8(+) T cells. Tumor-infiltrating lymphocytes from human mucosal lung cancer also expressed CD49a, which supports the relevance and possible extrapolation of these results in humans. We thus identified a link between the route of vaccination and the induction of a mucosal homing program on induced CD8(+) T cells that controlled their trafficking. Immunization route directly affected the efficacy of the cancer vaccine to control mucosal tumors.
in vivo CD8+ T cell depletion
Kearl, T. J., et al. (2013). "Programmed death receptor-1/programmed death receptor ligand-1 blockade after transient lymphodepletion to treat myeloma" J Immunol 190(11): 5620-5628. PubMed
Early phase clinical trials targeting the programmed death receptor-1/ligand-1 (PD-1/PD-L1) pathway to overcome tumor-mediated immunosuppression have reported promising results for a variety of cancers. This pathway appears to play an important role in the failure of immune reactivity to malignant plasma cells in multiple myeloma patients, as the tumor cells express relatively high levels of PD-L1, and T cells show increased PD-1 expression. In the current study, we demonstrate that PD-1/PD-L1 blockade with a PD-L1-specific Ab elicits rejection of a murine myeloma when combined with lymphodepleting irradiation. This particular combined approach by itself has not previously been shown to be efficacious in other tumor models. The antitumor effect of lymphodepletion/anti-PD-L1 therapy was most robust when tumor Ag-experienced T cells were present either through cell transfer or survival after nonmyeloablative irradiation. In vivo depletion of CD4 or CD8 T cells completely eliminated antitumor efficacy of the lymphodepletion/anti-PD-L1 therapy, indicating that both T cell subsets are necessary for tumor rejection. Elimination of myeloma by T cells occurs relatively quickly as tumor cells in the bone marrow were nearly nondetectable by 5 d after the first anti-PD-L1 treatment, suggesting that antimyeloma reactivity is primarily mediated by preactivated T cells, rather than newly generated myeloma-reactive T cells. Anti-PD-L1 plus lymphodepletion failed to improve survival in two solid tumor models, but demonstrated significant efficacy in two hematologic malignancy models. In summary, our results support the clinical testing of lymphodepletion and PD-1/PD-L1 blockade as a novel approach for improving the survival of patients with multiple myeloma.
in vivo CD8+ T cell depletion
Pasche, N., et al. (2012). "The antibody-based delivery of interleukin-12 to the tumor neovasculature eradicates murine models of cancer in combination with paclitaxel" Clin Cancer Res 18(15): 4092-4103. PubMed
PURPOSE: Interleukin-12 (IL12) is a potent proinflammatory cytokine with antitumor activity. Its heterodimeric nature makes it compatible with a large variety of different immunocytokine formats. Here we report the design, production, and characterization of a novel immunocytokine, based on the fusion of the F8 antibody (specific to the alternatively spliced EDA domain of fibronectin, a marker of tumor neovasculature) with IL12 (termed IL12-F8-F8). EXPERIMENTAL DESIGN: We developed a novel immunocytokine based on the sequential fusion of interleukin-12 as a single polypeptide with two F8 antibodies in single-chain Fv (scFv) format. The fusion protein was characterized in vitro, and its targeting performance was assessed in vivo. The immunocytokine antitumor activity was studied as monotherapy as well as in combination therapies in three different murine tumor models. Moreover, depletion experiments and tumor analysis revealed a dominant role of natural killer cells for the mechanism of action. RESULTS: IL12-F8-F8 can be produced in mammalian cells, yielding a product of good pharmaceutical quality, capable of selective localization on the tumor neovasculature in vivo, as judged by quantitative biodistribution analysis with radioiodinated protein preparations. The protein potently inhibited tumor growth in three different immunocompetent syngeneic models of cancer. The treatment was generally well tolerated. Moreover, the IL12-F8-F8 fusion protein could be produced both with murine IL12 (mIL12) and with human IL12 (hIL12). CONCLUSIONS: The potent antitumor activity of mIL12-F8-F8, studied alone or in combination with paclitaxel in different tumor models, paves the way to the clinical development of the fully human immunocytokine.
in vivo CD8+ T cell depletion
Quezada, S. A., et al. (2010). "Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts" J Exp Med 207(3): 637-650. PubMed
Adoptive transfer of large numbers of tumor-reactive CD8(+) cytotoxic T lymphocytes (CTLs) expanded and differentiated in vitro has shown promising clinical activity against cancer. However, such protocols are complicated by extensive ex vivo manipulations of tumor-reactive cells and have largely focused on CD8(+) CTLs, with much less emphasis on the role and contribution of CD4(+) T cells. Using a mouse model of advanced melanoma, we found that transfer of small numbers of naive tumor-reactive CD4(+) T cells into lymphopenic recipients induces substantial T cell expansion, differentiation, and regression of large established tumors without the need for in vitro manipulation. Surprisingly, CD4(+) T cells developed cytotoxic activity, and tumor rejection was dependent on class II-restricted recognition of tumors by tumor-reactive CD4(+) T cells. Furthermore, blockade of the coinhibitory receptor CTL-associated antigen 4 (CTLA-4) on the transferred CD4(+) T cells resulted in greater expansion of effector T cells, diminished accumulation of tumor-reactive regulatory T cells, and superior antitumor activity capable of inducing regression of spontaneous mouse melanoma. These findings suggest a novel potential therapeutic role for cytotoxic CD4(+) T cells and CTLA-4 blockade in cancer immunotherapy, and demonstrate the potential advantages of differentiating tumor-reactive CD4(+) cells in vivo over current protocols favoring in vitro expansion and differentiation.