InVivoPlus anti-mouse IL-10R (CD210)
Product Details
The 1B1.3A monoclonal antibody reacts with mouse IL-10R (IL-10 receptor) also known as CD210. The IL-10R is a class II cytokine receptor and is expressed by a variety of cell types including thymocytes, T lymphocytes, B lymphocytes, NK cells, monocytes, and macrophages. Upon binding IL-10, IL-10R stimulation results in many pleiotropic, effects in immunoregulation and inflammation. IL-10R downregulates the expression of pro-inflammatory cytokines, MHC class II antigens, and co-stimulatory molecules on macrophages. It also enhances B lymphocyte survival, proliferation, and antibody production. IL-10R signaling can block NF-κB activity, and is involved in the regulation of the JAK-STAT signaling pathway. The 1B1.3A antibody is a neutralizing antibody and has been shown to block the binding of human IL-10, which cross-reacts with the mouse IL-10R. However, this clone does not recognize the human IL-10R.Specifications
| Isotype | Rat IgG1, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoPlus rat IgG1 isotype control, anti-horseradish peroxidase |
| Recommended Dilution Buffer | InVivoPure pH 6.5T Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Recombinant ligand-binding domain of mouse IL-10R |
| Reported Applications |
in vivo blocking of IL-10/IL-10R signaling in vitro blocking of IL-10R signaling Flow cytometry Western Blot |
| Formulation |
PBS, pH 6.5 0.01% Tween Contains no stabilizers or preservatives |
| Endotoxin* |
<1EU/mg (<0.001EU/μg) Determined by LAL gel clotting assay |
| Aggregation* |
<5% Determined by SEC |
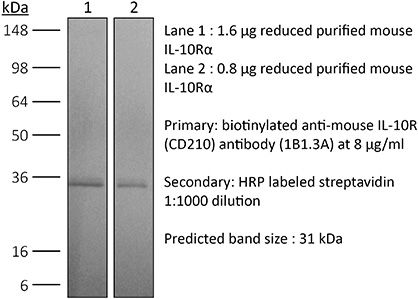
| Purity |
>95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_1107611 |
| Molecular Weight | 150 kDa |
| Murine Pathogen Tests* |
Ectromelia/Mousepox Virus: Negative Hantavirus: Negative K Virus: Negative Lactate Dehydrogenase-Elevating Virus: Negative Lymphocytic Choriomeningitis virus: Negative Mouse Adenovirus: Negative Mouse Cytomegalovirus: Negative Mouse Hepatitis Virus: Negative Mouse Minute Virus: Negative Mouse Norovirus: Negative Mouse Parvovirus: Negative Mouse Rotavirus: Negative Mycoplasma Pulmonis: Negative Pneumonia Virus of Mice: Negative Polyoma Virus: Negative Reovirus Screen: Negative Sendai Virus: Negative Theiler’s Murine Encephalomyelitis: Negative |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
Additional Formats

Recommended Products
-

Recommended Isotype Control(s)
InVivoPlus rat IgG1 isotype control, anti-horseradish peroxidase
-

Recommended Dilution Buffer
InVivoPure pH 6.5T Dilution Buffer
in vivo blocking of IL-10/IL-10R signaling
Xu, M., et al. (2018). "c-MAF-dependent regulatory T cells mediate immunological tolerance to a gut pathobiont" Nature 554(7692): 373-377. PubMed
Both microbial and host genetic factors contribute to the pathogenesis of autoimmune diseases. There is accumulating evidence that microbial species that potentiate chronic inflammation, as in inflammatory bowel disease, often also colonize healthy individuals. These microorganisms, including the Helicobacter species, can induce pathogenic T cells and are collectively referred to as pathobionts. However, how such T cells are constrained in healthy individuals is not yet understood. Here we report that host tolerance to a potentially pathogenic bacterium, Helicobacter hepaticus, is mediated by the induction of RORgammat(+)FOXP3(+) regulatory T (iTreg) cells that selectively restrain pro-inflammatory T helper 17 (TH17) cells and whose function is dependent on the transcription factor c-MAF. Whereas colonization of wild-type mice by H. hepaticus promoted differentiation of RORgammat-expressing microorganism-specific iTreg cells in the large intestine, in disease-susceptible IL-10-deficient mice, there was instead expansion of colitogenic TH17 cells. Inactivation of c-MAF in the Treg cell compartment impaired differentiation and function, including IL-10 production, of bacteria-specific iTreg cells, and resulted in the accumulation of H. hepaticus-specific inflammatory TH17 cells and spontaneous colitis. By contrast, RORgammat inactivation in Treg cells had only a minor effect on the bacteria-specific Treg and TH17 cell balance, and did not result in inflammation. Our results suggest that pathobiont-dependent inflammatory bowel disease is driven by microbiota-reactive T cells that have escaped this c-MAF-dependent mechanism of iTreg-TH17 homeostasis.
in vivo blocking of IL-10/IL-10R signaling
Burrack, K. S., et al. (2018). "Interleukin-15 Complex Treatment Protects Mice from Cerebral Malaria by Inducing Interleukin-10-Producing Natural Killer Cells" Immunity 48(4): 760-772 e764. PubMed
Cerebral malaria is a deadly complication of Plasmodium infection and involves blood brain barrier (BBB) disruption following infiltration of white blood cells. During experimental cerebral malaria (ECM), mice inoculated with Plasmodium berghei ANKA-infected red blood cells develop a fatal CM-like disease caused by CD8(+) T cell-mediated pathology. We found that treatment with interleukin-15 complex (IL-15C) prevented ECM, whereas IL-2C treatment had no effect. IL-15C-expanded natural killer (NK) cells were necessary and sufficient for protection against ECM. IL-15C treatment also decreased CD8(+) T cell activation in the brain and prevented BBB breakdown without influencing parasite load. IL-15C induced NK cells to express IL-10, which was required for IL-15C-mediated protection against ECM. Finally, we show that ALT-803, a modified human IL-15C, mediates similar induction of IL-10 in NK cells and protection against ECM. These data identify a regulatory role for cytokine-stimulated NK cells in the prevention of a pathogenic immune response.
in vivo blocking of IL-10/IL-10R signaling
Sun, M., et al. (2018). "Microbiota-derived short-chain fatty acids promote Th1 cell IL-10 production to maintain intestinal homeostasis" Nat Commun 9(1): 3555. PubMed
T-cells are crucial in maintanence of intestinal homeostasis, however, it is still unclear how microbiota metabolites regulate T-effector cells. Here we show gut microbiota-derived short-chain fatty acids (SCFAs) promote microbiota antigen-specific Th1 cell IL-10 production, mediated by G-protein coupled receptors 43 (GPR43). Microbiota antigen-specific Gpr43(-/-) CBir1 transgenic (Tg) Th1 cells, specific for microbiota antigen CBir1 flagellin, induce more severe colitis compared with wide type (WT) CBir1 Tg Th1 cells in Rag(-/-) recipient mice. Treatment with SCFAs limits colitis induction by promoting IL-10 production, and administration of anti-IL-10R antibody promotes colitis development. Mechanistically, SCFAs activate Th1 cell STAT3 and mTOR, and consequently upregulate transcription factor B lymphocyte-induced maturation protein 1 (Blimp-1), which mediates SCFA-induction of IL-10. SCFA-treated Blimp1(-/-) Th1 cells produce less IL-10 and induce more severe colitis compared to SCFA-treated WT Th1 cells. Our studies, thus, provide insight into how microbiota metabolites regulate Th1 cell functions to maintain intestinal homeostasis.
in vivo blocking of IL-10/IL-10R signaling
Christensen, A. D., et al. (2015). "Depletion of regulatory T cells in a hapten-induced inflammation model results in prolonged and increased inflammation driven by T cells" Clin Exp Immunol 179(3): 485-499. PubMed
Regulatory T cells (Tregs ) are known to play an immunosuppressive role in the response of contact hypersensitivity (CHS), but neither the dynamics of Tregs during the CHS response nor the exaggerated inflammatory response after depletion of Tregs has been characterized in detail. In this study we show that the number of Tregs in the challenged tissue peak at the same time as the ear-swelling reaches its maximum on day 1 after challenge, whereas the number of Tregs in the draining lymph nodes peaks at day 2. As expected, depletion of Tregs by injection of a monoclonal antibody to CD25 prior to sensitization led to a prolonged and sustained inflammatory response which was dependent upon CD8 T cells, and co-stimulatory blockade with cytotoxic T lymphocyte antigen-4-immunoglobulin (CTLA-4-Ig) suppressed the exaggerated inflammation. In contrast, blockade of the interleukin (IL)-10-receptor (IL-10R) did not further increase the exaggerated inflammatory response in the Treg -depleted mice. In the absence of Tregs , the response changed from a mainly acute reaction with heavy infiltration of neutrophils to a sustained response with more chronic characteristics (fewer neutrophils and dominated by macrophages). Furthermore, depletion of Tregs enhanced the release of cytokines and chemokines locally in the inflamed ear and augmented serum levels of the systemic inflammatory mediators serum amyloid (SAP) and haptoglobin early in the response.
in vivo blocking of IL-10/IL-10R signaling
Liu, G., et al. (2015). "IL-27 Signaling Is Crucial for Survival of Mice Infected with African Trypanosomes via Preventing Lethal Effects of CD4+ T Cells and IFN-gamma" PLoS Pathog 11(7): e1005065. PubMed
African trypanosomes are extracellular protozoan parasites causing a chronic debilitating disease associated with a persistent inflammatory response. Maintaining the balance of the inflammatory response via downregulation of activation of M1-type myeloid cells was previously shown to be crucial to allow prolonged survival. Here we demonstrate that infection with African trypanosomes of IL-27 receptor-deficient (IL-27R-/-) mice results in severe liver immunopathology and dramatically reduced survival as compared to wild-type mice. This coincides with the development of an exacerbated Th1-mediated immune response with overactivation of CD4+ T cells and strongly enhanced production of inflammatory cytokines including IFN-gamma. What is important is that IL-10 production was not impaired in infected IL-27R-/- mice. Depletion of CD4+ T cells in infected IL-27R-/- mice resulted in a dramatically reduced production of IFN-gamma, preventing the early mortality of infected IL-27R-/- mice. This was accompanied by a significantly reduced inflammatory response and a major amelioration of liver pathology. These results could be mimicked by treating IL-27R-/- mice with a neutralizing anti-IFN-gamma antibody. Thus, our data identify IL-27 signaling as a novel pathway to prevent early mortality via inhibiting hyperactivation of CD4+ Th1 cells and their excessive secretion of IFN-gamma during infection with African trypanosomes. These data are the first to demonstrate the essential role of IL-27 signaling in regulating immune responses to extracellular protozoan infections.
in vivo blocking of IL-10/IL-10R signaling
Lin, C. C., et al. (2014). "Bhlhe40 controls cytokine production by T cells and is essential for pathogenicity in autoimmune neuroinflammation" Nat Commun 5: 3551. PubMed
TH1 and TH17 cells mediate neuroinflammation in experimental autoimmune encephalomyelitis (EAE), a mouse model of multiple sclerosis. Pathogenic TH cells in EAE must produce the pro-inflammatory cytokine granulocyte-macrophage colony stimulating factor (GM-CSF). TH cell pathogenicity in EAE is also regulated by cell-intrinsic production of the immunosuppressive cytokine interleukin 10 (IL-10). Here we demonstrate that mice deficient for the basic helix-loop-helix (bHLH) transcription factor Bhlhe40 (Bhlhe40(-/-)) are resistant to the induction of EAE. Bhlhe40 is required in vivo in a T cell-intrinsic manner, where it positively regulates the production of GM-CSF and negatively regulates the production of IL-10. In vitro, GM-CSF secretion is selectively abrogated in polarized Bhlhe40(-/-) TH1 and TH17 cells, and these cells show increased production of IL-10. Blockade of IL-10 receptor in Bhlhe40(-/-) mice renders them susceptible to EAE. These findings identify Bhlhe40 as a critical regulator of autoreactive T-cell pathogenicity.
in vitro blocking of IL-10R signaling
Verhagen, J. and D. C. Wraith. (2014). "Blockade of LFA-1 augments in vitro differentiation of antigen-induced Foxp3(+) Treg cells" J Immunol Methods 414: 58-64. PubMed
Adoptive transfer of antigen-specific, in vitro-induced Foxp3(+) Treg (iTreg) cells protects against autoimmune disease. To generate antigen-specific iTreg cells at high purity, however, remains a challenge. Whereas polyclonal T cell stimulation with anti-CD3 and anti-CD28 antibody yields Foxp3(+) iTreg cells at a purity of 90-95%, antigen-induced iTreg cells typically do not exceed a purity of 65-75%, even in a TCR-transgenic model. In a similar vein to thymic Treg cell selection, iTreg cell differentiation is influenced not only by antigen recognition and the availability of TGF-beta but also by co-factors including costimulation and adhesion molecules. In this study, we demonstrate that blockade of the T cell integrin Leukocyte Function-associated Antigen-1 (LFA-1) during antigen-mediated iTreg cell differentiation augments Foxp3 induction, leading to approximately 90% purity of Foxp3(+) iTreg cells. This increased efficacy not only boosts the yield of Foxp3(+) iTreg cells, it also reduces contamination with activated effector T cells, thus improving the safety of adoptive transfer immunotherapy.
in vivo blocking of IL-10/IL-10R signaling
Masson, F., et al. (2014). "Id2 represses E2A-mediated activation of IL-10 expression in T cells" Blood 123(22): 3420-3428. PubMed
Interleukin-10 (IL-10) is a key immunoregulatory cytokine that functions to prevent inflammatory and autoimmune diseases. Despite the critical role for IL-10 produced by effector CD8(+) T cells during pathogen infection and autoimmunity, the mechanisms regulating its production are poorly understood. We show that loss of the inhibitor of DNA binding 2 (Id2) in T cells resulted in aberrant IL-10 expression in vitro and in vivo during influenza virus infection and in a model of acute graft-versus-host disease (GVHD). Furthermore, IL-10 overproduction substantially reduced the immunopathology associated with GVHD. We demonstrate that Id2 acts to repress the E2A-mediated trans-activation of the Il10 locus. Collectively, our findings uncover a key regulatory role of Id2 during effector T cell differentiation necessary to limit IL-10 production by activated T cells and minimize their suppressive activity during the effector phase of disease control.
in vivo blocking of IL-10/IL-10R signaling
Dolina, J. S., et al. (2014). "Liver-primed CD8+ T cells suppress antiviral adaptive immunity through galectin-9-independent T-cell immunoglobulin and mucin 3 engagement of high-mobility group box 1 in mice" Hepatology 59(4): 1351-1365. PubMed
The liver is a tolerogenic environment exploited by persistent infections, such as hepatitis B (HBV) and C (HCV) viruses. In a murine model of intravenous hepatotropic adenovirus infection, liver-primed antiviral CD8(+) T cells fail to produce proinflammatory cytokines and do not display cytolytic activity characteristic of effector CD8(+) T cells generated by infection at an extrahepatic, that is, subcutaneous, site. Importantly, liver-generated CD8(+) T cells also appear to have a T-regulatory (Treg) cell function exemplified by their ability to limit proliferation of antigen-specific T-effector (Teff ) cells in vitro and in vivo via T-cell immunoglobulin and mucin 3 (Tim-3) expressed by the CD8(+) Treg cells. Regulatory activity did not require recognition of the canonical Tim-3 ligand, galectin-9, but was dependent on CD8(+) Treg cell-surface Tim-3 binding to the alarmin, high-mobility group box 1 (HMGB-1). CONCLUSION: Virus-specific Tim-3(+) CD8(+) T cells operating through HMGB-1 recognition in the setting of acute and chronic viral infections of the liver may act to dampen hepatic T-cell responses in the liver microenvironment and, as a consequence, limit immune-mediated tissue injury or promote the establishment of persistent infections.
in vivo blocking of IL-10/IL-10R signaling
Ruffell, B., et al. (2014). "Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells" Cancer Cell 26(5): 623-637. PubMed
Blockade of colony-stimulating factor-1 (CSF-1) limits macrophage infiltration and improves response of mammary carcinomas to chemotherapy. Herein we identify interleukin (IL)-10 expression by macrophages as the critical mediator of this phenotype. Infiltrating macrophages were the primary source of IL-10 within tumors, and therapeutic blockade of IL-10 receptor (IL-10R) was equivalent to CSF-1 neutralization in enhancing primary tumor response to paclitaxel and carboplatin. Improved response to chemotherapy was CD8(+) T cell-dependent, but IL-10 did not directly suppress CD8(+) T cells or alter macrophage polarization. Instead, IL-10R blockade increased intratumoral dendritic cell expression of IL-12, which was necessary for improved outcomes. In human breast cancer, expression of IL12A and cytotoxic effector molecules were predictive of pathological complete response rates to paclitaxel.
in vivo blocking of IL-10/IL-10R signaling
Richter, K. and A. Oxenius. (2013). "Non-neutralizing antibodies protect from chronic LCMV infection independently of activating FcgammaR or complement" Eur J Immunol 43(9): 2349-2360. PubMed
Chronic viral infections lead to CD8(+) T cell exhaustion, characterized by impaired cytokine secretion. The presence of the immune-regulatory cytokine IL-10 promotes chronic lymphocytic choriomeningitis virus (LCMV) Clone 13 infection in mice, whereas the absence of IL-10/IL-10R signaling early during infection results in viral clearance and higher percentages and numbers of antiviral, cytokine-producing T cells. However, it is currently unclear which cell populations and effector molecules are crucial to protect against chronic infection. In this study, we demonstrate that antiviral, LCMV-binding, non-neutralizing antibodies are needed, in addition to CD4(+) and CD8(+) T cells, to clear a high-dose LCMV infection in mice, in the absence of IL-10. The interaction between CD4(+) T cells and B cells in B-cell follicles via CD40/CD40L, in addition to class switch and/or somatic hypermutation, is crucial for viral control in the absence of IL-10. Interestingly, transfer of LCMV-binding non-neutralizing antibodies protected recipients from chronic infection. In addition, viral clearance in the absence of IL-10R signaling was independent of activating Fcgamma receptors and complement. These data highlight that non-neutralizing antibodies effectively contribute to the control of LCMV infection when present prior to infection, suggesting that the induction of neutralizing antibodies is not implicitly necessary for the generation of successful vaccines.
in vivo blocking of IL-10/IL-10R signaling
Mishra, P. K., et al. (2013). "Prevention of type 1 diabetes through infection with an intestinal nematode parasite requires IL-10 in the absence of a Th2-type response" Mucosal Immunol 6(2): 297-308. PubMed
Helminth infection can prevent type 1 diabetes (T1D); however, the regulatory mechanisms inhibiting disease remain largely undefined. In these studies, nonobese diabetic (NOD) IL-4(-/-) mice were infected with the strictly enteric nematode parasite, Heligmosomoides polygyrus. Short-term infection, 5-7 weeks of age, inhibited T1D onset, as late as 40 weeks of age. CD4(+) T-cell STAT6 phosphorylation was inhibited, while suppressed signal transducer and activator of transcription 1 phosphorylation was sustained, as were increases in FOXP3(-), CD4(+) T-cell interleukin (IL)-10 production. Blockade of IL-10 signaling in NOD-IL-4(-/-), but not in NOD, mice during this short interval abrogated protective effects resulting in pancreatic beta-cell destruction and ultimately T1D. Transfer of CD4(+) T cells from H. polygyrus (Hp)-inoculated NOD IL-4(-/-) mice to NOD mice blocked the onset of T1D. These studies indicate that Hp infection induces non-T-regulatory cells to produce IL-10 independently of STAT6 signaling and that in this Th2-deficient environment IL-10 is essential for T1D inhibition.
in vitro blocking of IL-10R signaling, Flow Cytometry
Hu, Z., et al. (2013). "Regulatory CD8+ T cells associated with erosion of immune surveillance in persistent virus infection suppress in vitro and have a reversible proliferative defect" J Immunol 191(1): 312-322. PubMed
CD4(+) T cell help is critical for CD8(+) T cell memory and immune surveillance against persistent virus infections. Our recent data have showed the lack of CD4(+) T cells leads to the generation of an IL-10-producing CD8(+) T cell population during persistent murine gamma-herpesvirus-68 (MHV-68) infection. IL-10 from these cells is partly responsible for erosion in immune surveillance, leading to spontaneous virus reactivation in lungs. In this study, we further characterized the generation, phenotype, and function of these IL-10-producing CD8(+) T cells by comparing with a newly identified IL-10-producing CD8(+) T cell population present during the acute stage of the infection. The IL-10-producing CD8(+) populations in acute and chronic stages differed in their requirement for CD4(+) T cell help, the dependence on IL-2/CD25 and CD40-CD40L pathways, and the ability to proliferate in vitro in response to anti-CD3 stimulation. IL-10-producing CD8(+) T cells in the chronic stage showed a distinct immunophenotypic profile, sharing partial overlap with the markers of previously reported regulatory CD8(+) T cells, and suppressed the proliferation of naive CD8(+) T cells. Notably, they retained the ability to produce effector cytokines and cytotoxic activity. In addition, the proliferative defect of the cells could be restored by addition of exogenous IL-2 or blockade of IL-10. These data suggest that the IL-10-producing CD8(+) T cells arising in chronic MHV-68 infection in the absence of CD4(+) T cell help belong to a subset of CD8(+) regulatory T cells.
in vivo blocking of IL-10/IL-10R signaling
Wilson, M. S., et al. (2011). "IL-10 blocks the development of resistance to re-infection with Schistosoma mansoni" PLoS Pathog 7(8): e1002171. PubMed
Despite effective chemotherapy to treat schistosome infections, re-infection rates are extremely high. Resistance to reinfection can develop, however it typically takes several years following numerous rounds of treatment and re-infection, and often develops in only a small cohort of individuals. Using a well-established and highly permissive mouse model, we investigated whether immunoregulatory mechanisms influence the development of resistance. Following Praziquantel (PZQ) treatment of S. mansoni infected mice we observed a significant and mixed anti-worm response, characterized by Th1, Th2 and Th17 responses. Despite the elevated anti-worm response in PBMC’s, liver, spleen and mesenteric lymph nodes, this did not confer any protection from a secondary challenge infection. Because a significant increase in IL-10-producing CD4(+)CD44(+)CD25(+)GITR(+) lymphocytes was observed, we hypothesised that IL-10 was obstructing the development of resistance. Blockade of IL-10 combined with PZQ treatment afforded a greater than 50% reduction in parasite establishment during reinfection, compared to PZQ treatment alone, indicating that IL-10 obstructs the development of acquired resistance. Markedly enhanced Th1, Th2 and Th17 responses, worm-specific IgG1, IgG2b and IgE and circulating eosinophils characterized the protection. This study demonstrates that blocking IL-10 signalling during PZQ treatment can facilitate the development of protective immunity and provide a highly effective strategy to protect against reinfection with S. mansoni.
in vivo blocking of IL-10/IL-10R signaling
Kastenmuller, W., et al. (2011). "Regulatory T cells selectively control CD8+ T cell effector pool size via IL-2 restriction" J Immunol 187(6): 3186-3197. PubMed
Regulatory T cells (Treg) are key players in maintaining immune homeostasis but have also been shown to regulate immune responses against infectious pathogens. Therefore, Treg are a promising target for modulating immune responses to vaccines to improve their efficacy. Using a viral vector system, we found that Treg act on the developing immune response early postinfection by reducing the extent of dendritic cell costimulatory molecule expression. Due to this change and the lower IL-2 production that results, a substantial fraction of CD8(+) effector T cells lose CD25 expression several days after activation. Surprisingly, such Treg-dependent limitations in IL-2 signaling by Ag-activated CD8(+) T cells prevent effector differentiation without interfering with memory cell formation. In this way, Treg fine-tune the numbers of effector T cells generated while preserving the capacity for a rapid recall response upon pathogen re-exposure. This selective effect of Treg on a subpopulation of CD8(+) T cells indicates that although manipulation of the Treg compartment might not be optimal for prophylactic vaccinations, it can be potentially exploited to optimize vaccine efficacy for therapeutic interventions.