InVivoPlus anti-mouse CD40
Product Details
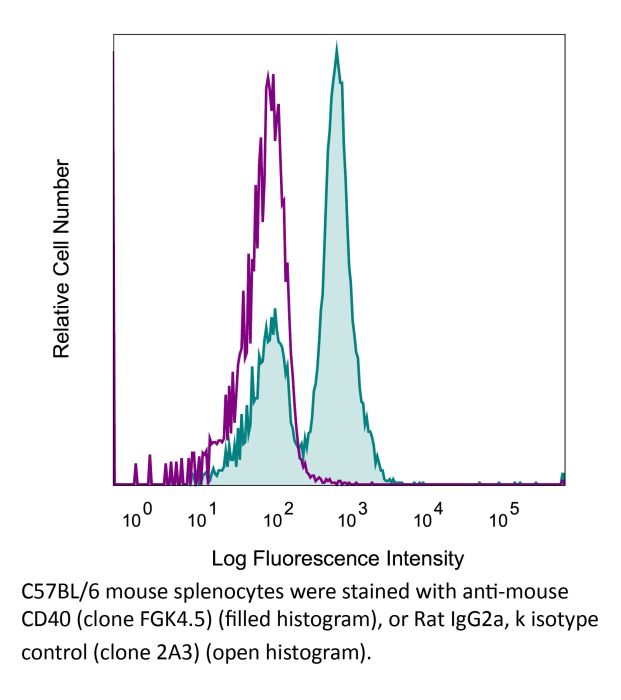
The FGK4.5 monoclonal antibody reacts with mouse CD40 also known as Bp50. CD40 is a 48 kDa type I transmembrane glycoprotein that belongs to the tumor necrosis factor receptor (TNFR) superfamily. CD40 is expressed broadly on antigen-presenting cells (APCs) such as dendritic cells, B cells, macrophages, and monocytes as well as non-immune endothelial cells, basal epithelial cells, and a range of tumors. Upon binding to its ligand CD154, CD40 acts as a costimulatory molecule for the activation of B cells, dendritic cells, monocytes, and other APCs. CD40 plays roles in B cell activation, differentiation, proliferation and Ig isotype switching as well as dendritic cell maturation. Agonistic CD40 monoclonal antibodies have been shown to activate APCs and promote anti-tumor T cell responses. The FGK4.5 antibody is an agonistic antibody that has been shown to activate CD40 expressing APCs. FGK4.5 can also be used to inhibit CD40/CD154 interaction in vitro and in vivo.Specifications
| Isotype | Rat IgG2a |
|---|---|
| Recommended Isotype Control(s) | InVivoPlus rat IgG2a isotype control, anti-trinitrophenol |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Recombinant mouse CD40 fusion protein |
| Reported Applications |
in vivo CD40 activation in vitro B cell stimulation/activation |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Aggregation* |
<5% Determined by SEC |
| Purity |
>95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_1107601 |
| Molecular Weight | 150 kDa |
| Murine Pathogen Tests* |
Ectromelia/Mousepox Virus: Negative Hantavirus: Negative K Virus: Negative Lactate Dehydrogenase-Elevating Virus: Negative Lymphocytic Choriomeningitis virus: Negative Mouse Adenovirus: Negative Mouse Cytomegalovirus: Negative Mouse Hepatitis Virus: Negative Mouse Minute Virus: Negative Mouse Norovirus: Negative Mouse Parvovirus: Negative Mouse Rotavirus: Negative Mycoplasma Pulmonis: Negative Pneumonia Virus of Mice: Negative Polyoma Virus: Negative Reovirus Screen: Negative Sendai Virus: Negative Theiler’s Murine Encephalomyelitis: Negative |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
Additional Formats

Recommended Products
-

Recommended Isotype Control(s)
InVivoPlus rat IgG2a isotype control, anti-trinitrophenol
-

Recommended Dilution Buffer
InVivoPure pH 7.0 Dilution Buffer
in vivo CD40 activation
Bauche, D., et al. (2018). "LAG3(+) Regulatory T Cells Restrain Interleukin-23-Producing CX3CR1(+) Gut-Resident Macrophages during Group 3 Innate Lymphoid Cell-Driven Colitis" Immunity 49(2): 342-352 e345. PubMed
Interleukin-22 (IL-22)-producing group 3 innate lymphoid cells (ILC3) maintains gut homeostasis but can also promote inflammatory bowel disease (IBD). The regulation of ILC3-dependent colitis remains to be elucidated. Here we show that Foxp3(+) regulatory T cells (Treg cells) prevented ILC3-mediated colitis in an IL-10-independent manner. Treg cells inhibited IL-23 and IL-1beta production from intestinal-resident CX3CR1(+) macrophages but not CD103(+) dendritic cells. Moreover, Treg cells restrained ILC3 production of IL-22 through suppression of CX3CR1(+) macrophage production of IL-23 and IL-1beta. This suppression was contact dependent and was mediated by latent activation gene-3 (LAG-3)-an immune checkpoint receptor-expressed on Treg cells. Engagement of LAG-3 on MHC class II drove profound immunosuppression of CX3CR1(+) tissue-resident macrophages. Our study reveals that the health of the intestinal mucosa is maintained by an axis driven by Treg cells communication with resident macrophages that withhold inflammatory stimuli required for ILC3 function.
in vivo CD40 activation
Carmi, Y., et al. (2015). "Allogeneic IgG combined with dendritic cell stimuli induce antitumour T-cell immunity" Nature 521(7550): 99-104. PubMed
Whereas cancers grow within host tissues and evade host immunity through immune-editing and immunosuppression, tumours are rarely transmissible between individuals. Much like transplanted allogeneic organs, allogeneic tumours are reliably rejected by host T cells, even when the tumour and host share the same major histocompatibility complex alleles, the most potent determinants of transplant rejection. How such tumour-eradicating immunity is initiated remains unknown, although elucidating this process could provide the basis for inducing similar responses against naturally arising tumours. Here we find that allogeneic tumour rejection is initiated in mice by naturally occurring tumour-binding IgG antibodies, which enable dendritic cells (DCs) to internalize tumour antigens and subsequently activate tumour-reactive T cells. We exploited this mechanism to treat autologous and autochthonous tumours successfully. Either systemic administration of DCs loaded with allogeneic-IgG-coated tumour cells or intratumoral injection of allogeneic IgG in combination with DC stimuli induced potent T-cell-mediated antitumour immune responses, resulting in tumour eradication in mouse models of melanoma, pancreas, lung and breast cancer. Moreover, this strategy led to eradication of distant tumours and metastases, as well as the injected primary tumours. To assess the clinical relevance of these findings, we studied antibodies and cells from patients with lung cancer. T cells from these patients responded vigorously to autologous tumour antigens after culture with allogeneic-IgG-loaded DCs, recapitulating our findings in mice. These results reveal that tumour-binding allogeneic IgG can induce powerful antitumour immunity that can be exploited for cancer immunotherapy.
in vivo CD40 activation
Conde, P., et al. (2015). "DC-SIGN(+) Macrophages Control the Induction of Transplantation Tolerance" Immunity 42(6): 1143-1158. PubMed
Tissue effector cells of the monocyte lineage can differentiate into different cell types with specific cell function depending on their environment. The phenotype, developmental requirements, and functional mechanisms of immune protective macrophages that mediate the induction of transplantation tolerance remain elusive. Here, we demonstrate that costimulatory blockade favored accumulation of DC-SIGN-expressing macrophages that inhibited CD8(+) T cell immunity and promoted CD4(+)Foxp3(+) Treg cell expansion in numbers. Mechanistically, that simultaneous DC-SIGN engagement by fucosylated ligands and TLR4 signaling was required for production of immunoregulatory IL-10 associated with prolonged allograft survival. Deletion of DC-SIGN-expressing macrophages in vivo, interfering with their CSF1-dependent development, or preventing the DC-SIGN signaling pathway abrogated tolerance. Together, the results provide new insights into the tolerogenic effects of costimulatory blockade and identify DC-SIGN(+) suppressive macrophages as crucial mediators of immunological tolerance with the concomitant therapeutic implications in the clinic.
in vitro B cell stimulation/activation
Xu, H., et al. (2015). "Regulation of bifurcating B cell trajectories by mutual antagonism between transcription factors IRF4 and IRF8" Nat Immunol . PubMed
Upon recognition of antigen, B cells undertake a bifurcated response in which some cells rapidly differentiate into plasmablasts while others undergo affinity maturation in germinal centers (GCs). Here we identified a double-negative feedback loop between the transcription factors IRF4 and IRF8 that regulated the initial developmental bifurcation of activated B cells as well as the GC response. IRF8 dampened signaling via the B cell antigen receptor (BCR), facilitated antigen-specific interaction with helper T cells, and promoted antibody affinity maturation while antagonizing IRF4-driven differentiation of plasmablasts. Genomic analysis revealed concentration-dependent actions of IRF4 and IRF8 in regulating distinct gene-expression programs. Stochastic modeling suggested that the double-negative feedback was sufficient to initiate bifurcation of the B cell developmental trajectories.
in vivo CD40 activation
Bartkowiak, T., et al. (2015). "Unique potential of 4-1BB agonist antibody to promote durable regression of HPV+ tumors when combined with an E6/E7 peptide vaccine" Proc Natl Acad Sci U S A 112(38): E5290-5299. PubMed
Antibody modulation of T-cell coinhibitory (e.g., CTLA-4) or costimulatory (e.g., 4-1BB) receptors promotes clinical responses to a variety of cancers. Therapeutic cancer vaccination, in contrast, has produced limited clinical benefit and no curative therapies. The E6 and E7 oncoproteins of human papilloma virus (HPV) drive the majority of genital cancers, and many oropharyngeal tumors. We discovered 15-19 amino acid peptides from HPV-16 E6/E7 for which induction of T-cell immunity correlates with disease-free survival in patients treated for high-grade cervical neoplasia. We report here that intranasal vaccination with these peptides and the adjuvant alpha-galactosylceramide elicits systemic and mucosal T-cell responses leading to reduced HPV(+) TC-1 tumor growth and prolonged survival in mice. We hypothesized that the inability of these T cells to fully reject established tumors resulted from suppression in the tumor microenvironment which could be ameliorated through checkpoint modulation. Combining this E6/E7 peptide vaccine with checkpoint blockade produced only modest benefit; however, coadministration with a 4-1BB agonist antibody promoted durable regression of established genital TC-1 tumors. Relative to other therapies tested, this combination of vaccine and alpha4-1BB promoted the highest CD8(+) versus regulatory FoxP3(+) T-cell ratios, elicited 2- to 5-fold higher infiltration by E7-specific CTL, and evoked higher densities of highly cytotoxic TcEO (T cytotoxic Eomesodermin) CD8 (>70-fold) and ThEO (T helper Eomesodermin) CD4 (>17-fold) T cells. These findings have immediate clinical relevance both in terms of the direct clinical utility of the vaccine studied and in illustrating the potential of 4-1BB antibody to convert therapeutic E6/E7 vaccines already in clinical trials into curative therapies.
in vivo CD40 activation
Rabenstein, H., et al. (2014). "Differential kinetics of antigen dependency of CD4+ and CD8+ T cells" J Immunol 192(8): 3507-3517. PubMed
Ag recognition via the TCR is necessary for the expansion of specific T cells that then contribute to adaptive immunity as effector and memory cells. Because CD4+ and CD8+ T cells differ in terms of their priming APCs and MHC ligands we compared their requirements of Ag persistence during their expansion phase side by side. Proliferation and effector differentiation of TCR transgenic and polyclonal mouse T cells were thus analyzed after transient and continuous TCR signals. Following equally strong stimulation, CD4+ T cell proliferation depended on prolonged Ag presence, whereas CD8+ T cells were able to divide and differentiate into effector cells despite discontinued Ag presentation. CD4+ T cell proliferation was neither affected by Th lineage or memory differentiation nor blocked by coinhibitory signals or missing inflammatory stimuli. Continued CD8+ T cell proliferation was truly independent of self-peptide/MHC-derived signals. The subset divergence was also illustrated by surprisingly broad transcriptional differences supporting a stronger propensity of CD8+ T cells to programmed expansion. These T cell data indicate an intrinsic difference between CD4+ and CD8+ T cells regarding the processing of TCR signals for proliferation. We also found that the presentation of a MHC class II-restricted peptide is more efficiently prolonged by dendritic cell activation in vivo than a class I bound one. In summary, our data demonstrate that CD4+ T cells require continuous stimulation for clonal expansion, whereas CD8+ T cells can divide following a much shorter TCR signal.
in vitro B cell stimulation/activation
Muppidi, J. R., et al. (2014). "Loss of signalling via Galpha13 in germinal centre B-cell-derived lymphoma" Nature 516(7530): 254-258. PubMed
Germinal centre B-cell-like diffuse large B-cell lymphoma (GCB-DLBCL) is a common malignancy, yet the signalling pathways that are deregulated and the factors leading to its systemic dissemination are poorly defined. Work in mice showed that sphingosine-1-phosphate receptor-2 (S1PR2), a Galpha12 and Galpha13 coupled receptor, promotes growth regulation and local confinement of germinal centre B cells. Recent deep sequencing studies of GCB-DLBCL have revealed mutations in many genes in this cancer, including in GNA13 (encoding Galpha13) and S1PR2 (refs 5,6, 7). Here we show, using in vitro and in vivo assays, that GCB-DLBCL-associated mutations occurring in S1PR2 frequently disrupt the receptor’s Akt and migration inhibitory functions. Galpha13-deficient mouse germinal centre B cells and human GCB-DLBCL cells were unable to suppress pAkt and migration in response to S1P, and Galpha13-deficient mice developed germinal centre B-cell-derived lymphoma. Germinal centre B cells, unlike most lymphocytes, are tightly confined in lymphoid organs and do not recirculate. Remarkably, deficiency in Galpha13, but not S1PR2, led to germinal centre B-cell dissemination into lymph and blood. GCB-DLBCL cell lines frequently carried mutations in the Galpha13 effector ARHGEF1, and Arhgef1 deficiency also led to germinal centre B-cell dissemination. The incomplete phenocopy of Galpha13- and S1PR2 deficiency led us to discover that P2RY8, an orphan receptor that is mutated in GCB-DLBCL and another germinal centre B-cell-derived malignancy, Burkitt’s lymphoma, also represses germinal centre B-cell growth and promotes confinement via Galpha13. These findings identify a Galpha13-dependent pathway that exerts dual actions in suppressing growth and blocking dissemination of germinal centre B cells that is frequently disrupted in germinal centre B-cell-derived lymphoma.
in vivo CD40 activation
Erickson, J. J., et al. (2014). "Programmed death-1 impairs secondary effector lung CD8(+) T cells during respiratory virus reinfection" J Immunol 193(10): 5108-5117. PubMed
Reinfections with respiratory viruses are common and cause significant clinical illness, yet precise mechanisms governing this susceptibility are ill defined. Lung Ag-specific CD8(+) T cells (T(CD8)) are impaired during acute viral lower respiratory infection by the inhibitory receptor programmed death-1 (PD-1). To determine whether PD-1 contributes to recurrent infection, we first established a model of reinfection by challenging B cell-deficient mice with human metapneumovirus (HMPV) several weeks after primary infection, and found that HMPV replicated to high titers in the lungs. A robust secondary effector lung TCD8 response was generated during reinfection, but these cells were more impaired and more highly expressed the inhibitory receptors PD-1, LAG-3, and 2B4 than primary T(CD8). In vitro blockade demonstrated that PD-1 was the dominant inhibitory receptor early after reinfection. In vivo therapeutic PD-1 blockade during HMPV reinfection restored lung T(CD8) effector functions (i.e., degranulation and cytokine production) and enhanced viral clearance. PD-1 also limited the protective efficacy of HMPV epitope-specific peptide vaccination and impaired lung T(CD8) during heterotypic influenza virus challenge infection. Our results indicate that PD-1 signaling may contribute to respiratory virus reinfection and evasion of vaccine-elicited immune responses. These results have important implications for the design of effective vaccines against respiratory viruses.
in vivo CD40 activation
Hailemichael, Y., et al. (2013). "Persistent antigen at vaccination sites induces tumor-specific CD8(+) T cell sequestration, dysfunction and deletion" Nat Med 19(4): 465-472. PubMed
To understand why cancer vaccine-induced T cells often do not eradicate tumors, we studied immune responses in mice vaccinated with gp100 melanoma peptide in incomplete Freund’s adjuvant (peptide/IFA), which is commonly used in clinical cancer vaccine trials. Peptide/IFA vaccination primed tumor-specific CD8(+) T cells, which accumulated not in tumors but rather at the persisting, antigen-rich vaccination site. Once there, primed T cells became dysfunctional and underwent antigen-driven, interferon-gamma (IFN-gamma)- and Fas ligand (FasL)-mediated apoptosis, resulting in hyporesponsiveness to subsequent vaccination. Provision of CD40-specific antibody, Toll-like receptor 7 (TLR7) agonist and interleukin-2 (IL-2) reduced T cell apoptosis but did not prevent vaccination-site sequestration. A nonpersisting vaccine formulation shifted T cell localization toward tumors, inducing superior antitumor activity while reducing systemic T cell dysfunction and promoting memory formation. These data show that persisting vaccine depots can induce specific T cell sequestration, dysfunction and deletion at vaccination sites; short-lived formulations may overcome these limitations and result in greater therapeutic efficacy of peptide-based cancer vaccines.
in vivo CD40 activation
Locatelli, G., et al. (2012). "Primary oligodendrocyte death does not elicit anti-CNS immunity" Nat Neurosci 15(4): 543-550. PubMed
Anti-myelin immunity is commonly thought to drive multiple sclerosis, yet the initial trigger of this autoreactivity remains elusive. One of the proposed factors for initiating this disease is the primary death of oligodendrocytes. To specifically test such oligodendrocyte death as a trigger for anti-CNS immunity, we inducibly killed oligodendrocytes in an in vivo mouse model. Strong microglia-macrophage activation followed oligodendrocyte death, and myelin components in draining lymph nodes made CNS antigens available to lymphocytes. However, even conditions favoring autoimmunity-bystander activation, removal of regulatory T cells, presence of myelin-reactive T cells and application of demyelinating antibodies-did not result in the development of CNS inflammation after oligodendrocyte death. In addition, this lack of reactivity was not mediated by enhanced myelin-specific tolerance. Thus, in contrast with previously reported impairments of oligodendrocyte physiology, diffuse oligodendrocyte death alone or in conjunction with immune activation does not trigger anti-CNS immunity.
in vivo CD40 activation
Kurche, J. S., et al. (2012). "Type I IFN-dependent T cell activation is mediated by IFN-dependent dendritic cell OX40 ligand expression and is independent of T cell IFNR expression" J Immunol 188(2): 585-593. PubMed
Type I IFNs are important for direct control of viral infection and generation of adaptive immune responses. Recently, direct stimulation of CD4(+) T cells via type I IFNR has been shown to be necessary for the formation of functional CD4(+) T cell responses. In contrast, we find that CD4(+) T cells do not require intrinsic type I IFN signals in response to combined TLR/anti-CD40 vaccination. Rather, the CD4 response is dependent on the expression of type I IFNR (IFNalphaR) on innate cells. Further, we find that dendritic cell (DC) expression of the TNF superfamily member OX40 ligand was dependent on type I IFN signaling in the DC, resulting in a reduced CD4(+) T cell response that could be substantially rescued by an agonistic Ab to the receptor OX40. Taken together, we show that the IFNalphaR dependence of the CD4(+) T cell response is accounted for exclusively by defects in DC activation.