InVivoMAb anti-mouse CD28
Product Details
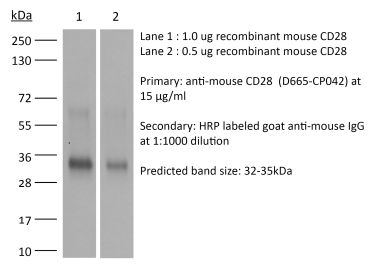
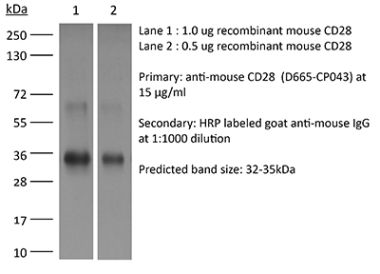
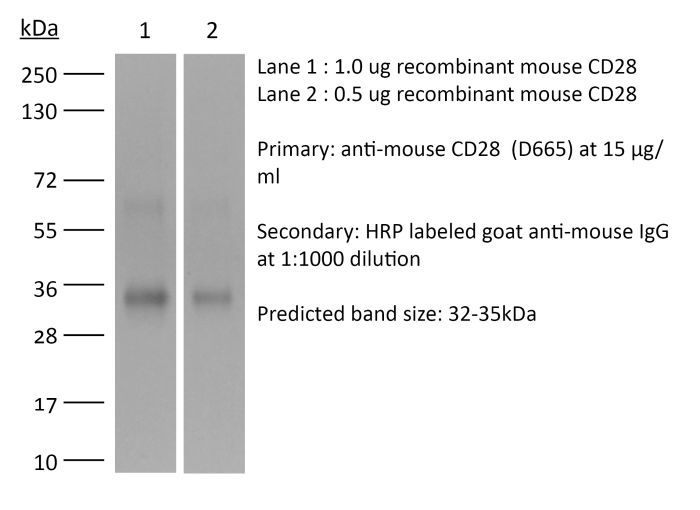
The D665 monoclonal antibody reacts with mouse CD28, a 45 kDa costimulatory receptor and a member of the Ig superfamily. CD28 is expressed by thymocytes, most peripheral T cells, and NK cells. CD28 is a receptor for CD80 (B7-1) and CD86 (B7-2). Signaling through CD28 induces IL-2 and IL-2 receptor expression and T cell proliferation. The D665 antibody is a CD28 superagonist and is most commonly used to induce the expansion of Treg cells in vivo in various mouse models of disease.Specifications
| Isotype | Mouse IgG1, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb mouse IgG1 isotype control, unknown specificity |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | A20 cells expressing mouse CD28 and a recombinant mouse CD28-Ig fusion protein |
| Reported Applications |
in vivo T cell stimulation/activation in vitro T cell stimulation/activation |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
<2EU/mg (<0.002EU/μg) Determined by LAL gel clotting assay |
| Purity |
>95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_2819055 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
Additional Formats


Recommended Products
-

Recommended Isotype Control(s)
InVivoMAb mouse IgG1 isotype control, unknown specificity
-

Recommended Dilution Buffer
InVivoPure pH 7.0 Dilution Buffer
in vivo T cell stimulation/activation
Win, S. J., et al. (2016). "In vivo activation of Treg cells with a CD28 superagonist prevents and ameliorates chronic destructive arthritis in mice" Eur J Immunol 46(5): 1193-1202. PubMed
Although regulatory T (Treg) cells are necessary to prevent autoimmune diseases, including arthritis, whether Treg cells can ameliorate established inflammatory disease is controversial. Using the glucose-6-phosphate isomerase (G6PI)-induced arthritis model in mice, we aimed to determine the therapeutic efficacy of increasing Treg cell number and function during chronic destructive arthritis. Chronic destructive arthritis was induced by transient depletion of Treg cells prior to immunization with G6PI. At different time points after disease induction, mice were treated with a CD28 superagonistic antibody (CD28SA). CD28SA treatment during the induction phase of arthritis ameliorated the acute signs of arthritis and completely prevented the development of chronic destructive arthritis. CD28SA treatment of mice with fully developed arthritis induced a significant reduction in clinical and histological signs of arthritis. When given during the chronic destructive phase of arthritis, 56 days after disease induction, CD28SA treatment resulted in a modest reduction of clinical signs of arthritis and a reduction in histopathological signs of joint inflammation. Our data show that increasing the number and activation of Treg cells by a CD28SA is therapeutically effective in experimental arthritis.
in vivo T cell stimulation/activation
Schmidt, T., et al. (2016). "Induction of T regulatory cells by the superagonistic anti-CD28 antibody D665 leads to decreased pathogenic IgG autoantibodies against desmoglein 3 in a HLA-transgenic mouse model of pemphigus vulgaris" Exp Dermatol 25(4): 293-298. PubMed
Pemphigus vulgaris (PV) is a potentially life-threatening autoimmune disease of the skin and mucous membranes. Its pathogenesis is based on IgG autoantibodies that target the desmosomal cadherins, desmoglein 3 (Dsg3) and desmoglein 1 (Dsg1) and induce intra-epidermal loss of adhesion. Although the PV pathogenesis is well-understood, therapeutic options are still limited to immunosuppressive drugs, particularly corticosteroids, which are associated with significant side effects. Dsg3-reactive T regulatory cells (Treg) have been previously identified in PV and healthy carriers of PV-associated HLA class II alleles. Ex vivo, Dsg3-specific Treg cells down-regulated the activation of pathogenic Dsg3-specific T-helper (Th) 2 cells. In this study, in a HLA-DRB1*04:02 transgenic mouse model of PV, peripheral Treg cells were modulated by the use of Treg-depleting or expanding monoclonal antibodies, respectively. Our findings show that, in vivo, although not statistically significant, Treg cells exert a clear down-regulatory effect on the Dsg3-driven T-cell response and, accordingly, the formation of Dsg3-specific IgG antibodies. These observations confirm the powerful immune regulatory functions of Treg cells and identify Treg cells as potential therapeutic modulators in PV.
in vivo T cell stimulation/activation
Schuhmann, M. K., et al. (2015). "CD28 superagonist-mediated boost of regulatory T cells increases thrombo-inflammation and ischemic neurodegeneration during the acute phase of experimental stroke" J Cereb Blood Flow Metab 35(1): 6-10. PubMed
While the detrimental role of non-regulatory T cells in ischemic stroke is meanwhile unequivocally recognized, there are controversies about the properties of regulatory T cells (Treg). The aim of this study was to elucidate the role of Treg by applying superagonistic anti-CD28 antibody expansion of Treg. Stroke outcome, thrombus formation, and brain-infiltrating cells were determined on day 1 after transient middle cerebral artery occlusion. Antibody-mediated expansion of Treg enhanced stroke size and worsened functional outcome. Mechanistically, Treg increased thrombus formation in the cerebral microvasculature. These findings confirm that Treg promote thrombo-inflammatory lesion growth during the acute stage of ischemic stroke.
in vitro T cell stimulation/activation
Dennehy, K. M., et al. (2006). "Cutting edge: monovalency of CD28 maintains the antigen dependence of T cell costimulatory responses" J Immunol 176(10): 5725-5729. PubMed
CD28 and CTLA-4 are the major costimulatory receptors on naive T cells. But it is not clear why CD28 is monovalent whereas CTLA-4 is bivalent for their shared ligands CD80/86. We generated bivalent CD28 constructs by fusing the extracellular domains of CTLA-4 or CD80 with the intracellular domains of CD28. Bivalent or monovalent CD28 constructs were ligated with recombinant ligands with or without TCR coligation. Monovalent CD28 ligation did not induce responses unless the TCR was coligated. By contrast, bivalent CD28 ligation induced responses in the absence of TCR engagement. To extend these findings to primary cells, we used novel superagonistic and conventional CD28 Abs. Superagonistic Ab D665, but not conventional Ab E18, predominantly ligates CD28 bivalently at low CD28/Ab ratios and induces Ag-independent T cell proliferation. Monovalency of CD28 for its natural ligands is thus essential to provide costimulation without inducing responses in the absence of TCR engagement.