InVivoMAb anti-mouse Ly6G
Product Details
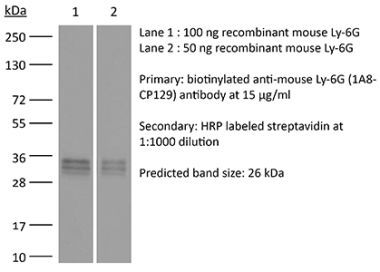
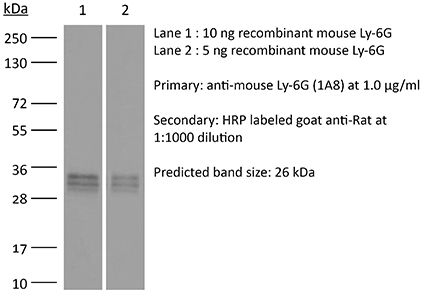
The 1A8 monoclonal antibody reacts with mouse Ly6G. Ly6G is a 21-25 kDa member of the Ly-6 superfamily of GPI-anchored cell surface proteins with roles in cell signaling and cell adhesion. Ly6G is expressed differentially during development by cells in the myeloid lineage including monocytes, macrophages, granulocytes, and neutrophils. Monocytes typically express Ly6G transiently during development while mature granulocytes and peripheral neutrophils retain expression making Ly6G a good cell surface marker for these populations. Unlike the RB6-8C5 antibody, the 1A8 antibody reacts specifically with mouse Ly6G with no reported cross reactivity with Ly6C.Specifications
| Isotype | Rat IgG2a, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb rat IgG2a isotype control, anti-trinitrophenol |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | EL4J cells transfected with Ly6G |
| Reported Applications |
in vivo neutrophil depletion in vivo MDSC depletion Immunofluorescence Immunohistochemistry (paraffin) Immunohistochemistry (frozen) Flow cytometry |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
<2EU/mg (<0.002EU/μg) Determined by LAL gel clotting assay |
| Purity |
>95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Purification | Protein G |
| RRID | AB_1107721 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
Additional Formats


Recommended Products
-

Recommended Isotype Control(s)
InVivoMAb rat IgG2a isotype control, anti-trinitrophenol
-

Recommended Dilution Buffer
InVivoPure pH 7.0 Dilution Buffer
in vivo neutrophil depletion
Davis, R. W. t., et al. (2018). "Luminol Chemiluminescence Reports Photodynamic Therapy-Generated Neutrophil Activity In Vivo and Serves as a Biomarker of Therapeutic Efficacy" Photochem Photobiol . PubMed
Inflammatory cells, most especially neutrophils, can be a necessary component of the antitumor activity occurring after administration of photodynamic therapy. Generation of neutrophil responses has been suggested to be particularly important in instances when the delivered photodynamic therapy (PDT) dose is insufficient. In these cases, the release of neutrophil granules and engagement of antitumor immunity may play an important role in eliminating residual disease. Herein, we utilize in vivo imaging of luminol chemiluminescence to noninvasively monitor neutrophil activation after PDT administration. Studies were performed in the AB12 murine model of mesothelioma, treated with Photofrin-PDT. Luminol-generated chemiluminescence increased transiently 1 h after PDT, followed by a subsequent decrease at 4 h after PDT. The production of luminol signal was not associated with the influx of Ly6G(+) cells, but was related to oxidative burst, as an indicator of neutrophil function. Most importantly, greater levels of luminol chemiluminescence 1 h after PDT were prognostic of a complete response at 90 days after PDT. Taken together, this research supports an important role for early activity by Ly6G(+) cells in the generation of long-term PDT responses in mesothelioma, and it points to luminol chemiluminescence as a potentially useful approach for preclinical monitoring of neutrophil activation by PDT.
in vivo neutrophil depletion
Moynihan, K. D., et al. (2016). "Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses" Nat Med. doi : 10.1038/nm.4200. PubMed
Checkpoint blockade with antibodies specific for cytotoxic T lymphocyte-associated protein (CTLA)-4 or programmed cell death 1 (PDCD1; also known as PD-1) elicits durable tumor regression in metastatic cancer, but these dramatic responses are confined to a minority of patients. This suboptimal outcome is probably due in part to the complex network of immunosuppressive pathways present in advanced tumors, which are unlikely to be overcome by intervention at a single signaling checkpoint. Here we describe a combination immunotherapy that recruits a variety of innate and adaptive immune cells to eliminate large tumor burdens in syngeneic tumor models and a genetically engineered mouse model of melanoma; to our knowledge tumors of this size have not previously been curable by treatments relying on endogenous immunity. Maximal antitumor efficacy required four components: a tumor-antigen-targeting antibody, a recombinant interleukin-2 with an extended half-life, anti-PD-1 and a powerful T cell vaccine. Depletion experiments revealed that CD8+ T cells, cross-presenting dendritic cells and several other innate immune cell subsets were required for tumor regression. Effective treatment induced infiltration of immune cells and production of inflammatory cytokines in the tumor, enhanced antibody-mediated tumor antigen uptake and promoted antigen spreading. These results demonstrate the capacity of an elicited endogenous immune response to destroy large, established tumors and elucidate essential characteristics of combination immunotherapies that are capable of curing a majority of tumors in experimental settings typically viewed as intractable.
in vivo neutrophil depletion
Conde, P., et al. (2015). "DC-SIGN(+) Macrophages Control the Induction of Transplantation Tolerance" Immunity 42(6): 1143-1158. PubMed
Tissue effector cells of the monocyte lineage can differentiate into different cell types with specific cell function depending on their environment. The phenotype, developmental requirements, and functional mechanisms of immune protective macrophages that mediate the induction of transplantation tolerance remain elusive. Here, we demonstrate that costimulatory blockade favored accumulation of DC-SIGN-expressing macrophages that inhibited CD8(+) T cell immunity and promoted CD4(+)Foxp3(+) Treg cell expansion in numbers. Mechanistically, that simultaneous DC-SIGN engagement by fucosylated ligands and TLR4 signaling was required for production of immunoregulatory IL-10 associated with prolonged allograft survival. Deletion of DC-SIGN-expressing macrophages in vivo, interfering with their CSF1-dependent development, or preventing the DC-SIGN signaling pathway abrogated tolerance. Together, the results provide new insights into the tolerogenic effects of costimulatory blockade and identify DC-SIGN(+) suppressive macrophages as crucial mediators of immunological tolerance with the concomitant therapeutic implications in the clinic.
in vivo neutrophil depletion
Griseri, T., et al. (2015). "Granulocyte Macrophage Colony-Stimulating Factor-Activated Eosinophils Promote Interleukin-23 Driven Chronic Colitis" Immunity 43(1): 187-199. PubMed
The role of intestinal eosinophils in immune homeostasis is enigmatic and the molecular signals that drive them from protective to tissue damaging are unknown. Most commonly associated with Th2 cell-mediated diseases, we describe a role for eosinophils as crucial effectors of the interleukin-23 (IL-23)-granulocyte macrophage colony-stimulating factor (GM-CSF) axis in colitis. Chronic intestinal inflammation was characterized by increased bone marrow eosinopoiesis and accumulation of activated intestinal eosinophils. IL-5 blockade or eosinophil depletion ameliorated colitis, implicating eosinophils in disease pathogenesis. GM-CSF was a potent activator of eosinophil effector functions and intestinal accumulation, and GM-CSF blockade inhibited chronic colitis. By contrast neutrophil accumulation was GM-CSF independent and dispensable for colitis. In addition to TNF secretion, release of eosinophil peroxidase promoted colitis identifying direct tissue-toxic mechanisms. Thus, eosinophils are key perpetrators of chronic inflammation and tissue damage in IL-23-mediated immune diseases and it suggests the GM-CSF-eosinophil axis as an attractive therapeutic target.
in vivo neutrophil depletion, Flow Cytometry, Immunohistochemistry (paraffin)
Coffelt, S. B., et al. (2015). "IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis" Nature 522(7556): 345-348. PubMed
Metastatic disease remains the primary cause of death for patients with breast cancer. The different steps of the metastatic cascade rely on reciprocal interactions between cancer cells and their microenvironment. Within this local microenvironment and in distant organs, immune cells and their mediators are known to facilitate metastasis formation. However, the precise contribution of tumour-induced systemic inflammation to metastasis and the mechanisms regulating systemic inflammation are poorly understood. Here we show that tumours maximize their chance of metastasizing by evoking a systemic inflammatory cascade in mouse models of spontaneous breast cancer metastasis. We mechanistically demonstrate that interleukin (IL)-1beta elicits IL-17 expression from gamma delta (gammadelta) T cells, resulting in systemic, granulocyte colony-stimulating factor (G-CSF)-dependent expansion and polarization of neutrophils in mice bearing mammary tumours. Tumour-induced neutrophils acquire the ability to suppress cytotoxic T lymphocytes carrying the CD8 antigen, which limit the establishment of metastases. Neutralization of IL-17 or G-CSF and absence of gammadelta T cells prevents neutrophil accumulation and downregulates the T-cell-suppressive phenotype of neutrophils. Moreover, the absence of gammadelta T cells or neutrophils profoundly reduces pulmonary and lymph node metastases without influencing primary tumour progression. Our data indicate that targeting this novel cancer-cell-initiated domino effect within the immune system–the gammadelta T cell/IL-17/neutrophil axis–represents a new strategy to inhibit metastatic disease.
in vivo neutrophil depletion, Flow Cytometry, Immunohistochemistry (paraffin), Immunohistochemistry (frozen)
Finisguerra, V., et al. (2015). "MET is required for the recruitment of anti-tumoural neutrophils" Nature 522(7556): 349-353. PubMed
Mutations or amplification of the MET proto-oncogene are involved in the pathogenesis of several tumours, which rely on the constitutive engagement of this pathway for their growth and survival. However, MET is expressed not only by cancer cells but also by tumour-associated stromal cells, although its precise role in this compartment is not well characterized. Here we show that MET is required for neutrophil chemoattraction and cytotoxicity in response to its ligand hepatocyte growth factor (HGF). Met deletion in mouse neutrophils enhances tumour growth and metastasis. This phenotype correlates with reduced neutrophil infiltration to both the primary tumour and metastatic sites. Similarly, Met is necessary for neutrophil transudation during colitis, skin rash or peritonitis. Mechanistically, Met is induced by tumour-derived tumour necrosis factor (TNF)-alpha or other inflammatory stimuli in both mouse and human neutrophils. This induction is instrumental for neutrophil transmigration across an activated endothelium and for inducible nitric oxide synthase production upon HGF stimulation. Consequently, HGF/MET-dependent nitric oxide release by neutrophils promotes cancer cell killing, which abates tumour growth and metastasis. After systemic administration of a MET kinase inhibitor, we prove that the therapeutic benefit of MET targeting in cancer cells is partly countered by the pro-tumoural effect arising from MET blockade in neutrophils. Our work identifies an unprecedented role of MET in neutrophils, suggests a potential ‘Achilles’ heel’ of MET-targeted therapies in cancer, and supports the rationale for evaluating anti-MET drugs in certain inflammatory diseases.
in vivo neutrophil depletion
Yamada, D. H., et al. (2015). "Suppression of Fcgamma-receptor-mediated antibody effector function during persistent viral infection" Immunity 42(2): 379-390. PubMed
Understanding how viruses subvert host immunity and persist is essential for developing strategies to eliminate infection. T cell exhaustion during chronic viral infection is well described, but effects on antibody-mediated effector activity are unclear. Herein, we show that increased amounts of immune complexes generated in mice persistently infected with lymphocytic choriomeningitis virus (LCMV) suppressed multiple Fcgamma-receptor (FcgammaR) functions. The high amounts of immune complexes suppressed antibody-mediated cell depletion, therapeutic antibody-killing of LCMV infected cells and human CD20-expressing tumors, as well as reduced immune complex-mediated cross-presentation to T cells. Suppression of FcgammaR activity was not due to inhibitory FcgammaRs or high concentrations of free antibody, and proper FcgammaR functions were restored when persistently infected mice specifically lacked immune complexes. Thus, we identify a mechanism of immunosuppression during viral persistence with implications for understanding effective antibody activity aimed at pathogen control.
in vivo neutrophil depletion
Ellis, G. T., et al. (2015). "TRAIL+ monocytes and monocyte-related cells cause lung damage and thereby increase susceptibility to influenza-Streptococcus pneumoniae coinfection" EMBO Rep 16(9): 1203-1218. PubMed
Streptococcus pneumoniae coinfection is a major cause of influenza-associated mortality; however, the mechanisms underlying pathogenesis or protection remain unclear. Using a clinically relevant mouse model, we identify immune-mediated damage early during coinfection as a new mechanism causing susceptibility. Coinfected CCR2(-/-) mice lacking monocytes and monocyte-derived cells control bacterial invasion better, show reduced epithelial damage and are overall more resistant than wild-type controls. In influenza-infected wild-type lungs, monocytes and monocyte-derived cells are the major cell populations expressing the apoptosis-inducing ligand TRAIL. Accordingly, anti-TRAIL treatment reduces bacterial load and protects against coinfection if administered during viral infection, but not following bacterial exposure. Post-influenza bacterial outgrowth induces a strong proinflammatory cytokine response and massive inflammatory cell infiltrate. Depletion of neutrophils or blockade of TNF-alpha facilitate bacterial outgrowth, leading to increased mortality, demonstrating that these factors aid bacterial control. We conclude that inflammatory monocytes recruited early, during the viral phase of coinfection, induce TRAIL-mediated lung damage, which facilitates bacterial invasion, while TNF-alpha and neutrophil responses help control subsequent bacterial outgrowth. We thus identify novel determinants of protection versus pathology in influenza-Streptococcus pneumoniae coinfection.
in vivo neutrophil depletion, Flow Cytometry
Moser, E. K., et al. (2014). "Late engagement of CD86 after influenza virus clearance promotes recovery in a FoxP3+ regulatory T cell dependent manner" PLoS Pathog 10(8): e1004315. PubMed
Influenza A virus (IAV) infection in the respiratory tract triggers robust innate and adaptive immune responses, resulting in both virus clearance and lung inflammation and injury. After virus clearance, resolution of ongoing inflammation and tissue repair occur during a distinct recovery period. B7 family co-stimulatory molecules such as CD80 and CD86 have important roles in modulating T cell activity during the initiation and effector stages of the host response to IAV infection, but their potential role during recovery and resolution of inflammation is unknown. We found that antibody-mediated CD86 blockade in vivo after virus clearance led to a delay in recovery, characterized by increased numbers of lung neutrophils and inflammatory cytokines in airways and lung interstitium, but no change in conventional IAV-specific T cell responses. However, CD86 blockade led to decreased numbers of FoxP3+ regulatory T cells (Tregs), and adoptive transfer of Tregs into alphaCD86 treated mice rescued the effect of the blockade, supporting a role for Tregs in promoting recovery after virus clearance. Specific depletion of Tregs late after infection mimicked the CD86 blockade phenotype, confirming a role for Tregs during recovery after virus clearance. Furthermore, we identified neutrophils as a target of Treg suppression since neutrophil depletion in Treg-depleted mice reduced excess inflammatory cytokines in the airways. These results demonstrate that Tregs, in a CD86 dependent mechanism, contribute to the resolution of disease after IAV infection, in part by suppressing neutrophil-driven cytokine release into the airways.
in vivo neutrophil depletion, Flow Cytometry
Chen, K. W., et al. (2014). "The neutrophil NLRC4 inflammasome selectively promotes IL-1beta maturation without pyroptosis during acute Salmonella challenge" Cell Rep 8(2): 570-582. PubMed
The macrophage NLRC4 inflammasome drives potent innate immune responses against Salmonella by eliciting caspase-1-dependent proinflammatory cytokine production (e.g., interleukin-1beta [IL-1beta]) and pyroptotic cell death. However, the potential contribution of other cell types to inflammasome-mediated host defense against Salmonella was unclear. Here, we demonstrate that neutrophils, typically viewed as cellular targets of IL-1beta, themselves activate the NLRC4 inflammasome during acute Salmonella infection and are a major cell compartment for IL-1beta production during acute peritoneal challenge in vivo. Importantly, unlike macrophages, neutrophils do not undergo pyroptosis upon NLRC4 inflammasome activation. The resistance of neutrophils to pyroptotic death is unique among inflammasome-signaling cells so far described and allows neutrophils to sustain IL-1beta production at a site of infection without compromising the crucial inflammasome-independent antimicrobial effector functions that would be lost if neutrophils rapidly lysed upon caspase-1 activation. Inflammasome pathway modification in neutrophils thus maximizes host proinflammatory and antimicrobial responses during pathogen challenge.
in vivo neutrophil depletion
Deshmukh, H. S., et al. (2014). "The microbiota regulates neutrophil homeostasis and host resistance to Escherichia coli K1 sepsis in neonatal mice" Nat Med 20(5): 524-530. PubMed
Neonatal colonization by microbes, which begins immediately after birth, is influenced by gestational age and the mother’s microbiota and is modified by exposure to antibiotics. In neonates, prolonged duration of antibiotic therapy is associated with increased risk of late-onset sepsis (LOS), a disorder controlled by neutrophils. A role for the microbiota in regulating neutrophil development and susceptibility to sepsis in the neonate remains unclear. We exposed pregnant mouse dams to antibiotics in drinking water to limit transfer of maternal microbes to the neonates. Antibiotic exposure of dams decreased the total number and composition of microbes in the intestine of the neonates. This was associated with decreased numbers of circulating and bone marrow neutrophils and granulocyte/macrophage-restricted progenitor cells in the bone marrow of antibiotic-treated and germ-free neonates. Antibiotic exposure of dams reduced the number of interleukin-17 (IL-17)-producing cells in the intestine and production of granulocyte colony-stimulating factor (G-CSF). Granulocytopenia was associated with impaired host defense and increased susceptibility to Escherichia coli K1 and Klebsiella pneumoniae sepsis in antibiotic-treated neonates, which could be partially reversed by administration of G-CSF. Transfer of a normal microbiota into antibiotic-treated neonates induced IL-17 production by group 3 innate lymphoid cells (ILCs) in the intestine, increasing plasma G-CSF levels and neutrophil numbers in a Toll-like receptor 4 (TLR4)- and myeloid differentiation factor 88 (MyD88)-dependent manner and restored IL-17-dependent resistance to sepsis. Specific depletion of ILCs prevented IL-17- and G-CSF-dependent granulocytosis and resistance to sepsis. These data support a role for the intestinal microbiota in regulation of granulocytosis, neutrophil homeostasis and host resistance to sepsis in neonates.
in vivo MDSC depletion
Deng, L., et al. (2014). "Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice" J Clin Invest 124(2): 687-695. PubMed
High-dose ionizing irradiation (IR) results in direct tumor cell death and augments tumor-specific immunity, which enhances tumor control both locally and distantly. Unfortunately, local relapses often occur following IR treatment, indicating that IR-induced responses are inadequate to maintain antitumor immunity. Therapeutic blockade of the T cell negative regulator programmed death-ligand 1 (PD-L1, also called B7-H1) can enhance T cell effector function when PD-L1 is expressed in chronically inflamed tissues and tumors. Here, we demonstrate that PD-L1 was upregulated in the tumor microenvironment after IR. Administration of anti-PD-L1 enhanced the efficacy of IR through a cytotoxic T cell-dependent mechanism. Concomitant with IR-mediated tumor regression, we observed that IR and anti-PD-L1 synergistically reduced the local accumulation of tumor-infiltrating myeloid-derived suppressor cells (MDSCs), which suppress T cells and alter the tumor immune microenvironment. Furthermore, activation of cytotoxic T cells with combination therapy mediated the reduction of MDSCs in tumors through the cytotoxic actions of TNF. Our data provide evidence for a close interaction between IR, T cells, and the PD-L1/PD-1 axis and establish a basis for the rational design of combination therapy with immune modulators and radiotherapy.
in vivo neutrophil depletion, Flow Cytometry, Immunohistochemistry (frozen)
Huang, L. R., et al. (2013). "Intrahepatic myeloid-cell aggregates enable local proliferation of CD8(+) T cells and successful immunotherapy against chronic viral liver infection" Nat Immunol 14(6): 574-583. PubMed
Chronic infection is difficult to overcome because of exhaustion or depletion of cytotoxic effector CD8(+) T cells (cytotoxic T lymphoytes (CTLs)). Here we report that signaling via Toll-like receptors (TLRs) induced intrahepatic aggregates of myeloid cells that enabled the population expansion of CTLs (iMATEs: ‘intrahepatic myeloid-cell aggregates for T cell population expansion’) without causing immunopathology. In the liver, CTL proliferation was restricted to iMATEs that were composed of inflammatory monocyte-derived CD11b(+) cells. Signaling via tumor-necrosis factor (TNF) caused iMATE formation that facilitated costimulation dependent on the receptor OX40 for expansion of the CTL population. The iMATEs arose during acute viral infection but were absent during chronic viral infection, yet they were still induced by TLR signaling. Such hepatic expansion of the CTL population controlled chronic viral infection of the liver after vaccination with DNA. Thus, iMATEs are dynamic structures that overcome regulatory cues that limit the population expansion of CTLs during chronic infection and can be used in new therapeutic vaccination strategies.
in vivo neutrophil depletion
Richter, K., et al. (2013). "Macrophage and T cell produced IL-10 promotes viral chronicity" PLoS Pathog 9(11): e1003735. PubMed
Chronic viral infections lead to CD8(+) T cell exhaustion, characterized by impaired cytokine secretion. Presence of the immune-regulatory cytokine IL-10 promotes chronicity of Lymphocytic Choriomeningitis Virus (LCMV) Clone 13 infection, while absence of IL-10/IL-10R signaling early during infection results in viral clearance and higher percentages and numbers of antiviral, cytokine producing T cells. IL-10 is produced by several cell types during LCMV infection but it is currently unclear which cellular sources are responsible for induction of viral chronicity. Here, we demonstrate that although dendritic cells produce IL-10 and overall IL-10 mRNA levels decrease significantly in absence of CD11c(+) cells, absence of IL-10 produced by CD11c(+) cells failed to improve the LCMV-specific T cell response and control of LCMV infection. Similarly, NK cell specific IL-10 deficiency had no positive impact on the LCMV-specific T cell response or viral control, even though high percentages of NK cells produced IL-10 at early time points after infection. Interestingly, we found markedly improved T cell responses and clearance of normally chronic LCMV Clone 13 infection when either myeloid cells or T cells lacked IL-10 production and mice depleted of monocytes/macrophages or CD4(+) T cells exhibited reduced overall levels of IL-10 mRNA. These data suggest that the decision whether LCMV infection becomes chronic or can be cleared critically depends on early CD4(+) T cell and monocyte/macrophage produced IL-10.
in vivo neutrophil depletion, Flow Cytometry
Garraud, K., et al. (2012). "Differential role of the interleukin-17 axis and neutrophils in resolution of inhalational anthrax" Infect Immun 80(1): 131-142. PubMed
The roles of interleukin-17 (IL-17) and neutrophils in the lung have been described as those of two intricate but independent players. Here we identify neutrophils as the primary IL-17-secreting subset of cells in a model of inhalation anthrax using A/J and C57BL/6 mice. With IL-17 receptor A knockout (IL-17RA-/-) mice, we confirmed that IL-17A/F signaling is instrumental in the self-recruitment of this population. We also show that the IL-17A/F axis is critical for surviving pulmonary infection, as IL-17RA-/- mice become susceptible to intranasal infection by Bacillus anthracis Sterne spores. Strikingly, infection with a fully virulent strain did not affect IL-17RA-/- mouse survival. Eventually, by depleting neutrophils in wild-type and IL-17RA-/- mice, we demonstrated the crucial role of IL-17-secreting neutrophils in mouse survival of infection by fully virulent B. anthracis. This work demonstrates the important roles of both IL-17 signaling and neutrophils in clearing this pathogen and surviving pulmonary B. anthracis infection.
in vivo neutrophil depletion, Flow Cytometry
Lee, W. B., et al. (2012). "Neutrophils Promote Mycobacterial Trehalose Dimycolate-Induced Lung Inflammation via the Mincle Pathway" PLoS Pathog 8(4): e1002614. PubMed
Trehalose 6,6′-dimycolate (TDM), a cord factor of Mycobacterium tuberculosis (Mtb), is an important regulator of immune responses during Mtb infections. Macrophages recognize TDM through the Mincle receptor and initiate TDM-induced inflammatory responses, leading to lung granuloma formation. Although various immune cells are recruited to lung granulomas, the roles of other immune cells, especially during the initial process of TDM-induced inflammation, are not clear. In this study, Mincle signaling on neutrophils played an important role in TDM-induced lung inflammation by promoting adhesion and innate immune responses. Neutrophils were recruited during the early stage of lung inflammation following TDM-induced granuloma formation. Mincle expression on neutrophils was required for infiltration of TDM-challenged sites in a granuloma model induced by TDM-coated-beads. TDM-induced Mincle signaling on neutrophils increased cell adherence by enhancing F-actin polymerization and CD11b/CD18 surface expression. The TDM-induced effects were dependent on Src, Syk, and MAPK/ERK kinases (MEK). Moreover, coactivation of the Mincle and TLR2 pathways by TDM and Pam3CSK4 treatment synergistically induced CD11b/CD18 surface expression, reactive oxygen species, and TNFalpha production by neutrophils. These synergistically-enhanced immune responses correlated with the degree of Mincle expression on neutrophil surfaces. The physiological relevance of the Mincle-mediated anti-TDM immune response was confirmed by defective immune responses in Mincle(-)/(-) mice upon aerosol infections with Mtb. Mincle-mutant mice had higher inflammation levels and mycobacterial loads than WT mice. Neutrophil depletion with anti-Ly6G antibody caused a reduction in IL-6 and monocyte chemotactic protein-1 expression upon TDM treatment, and reduced levels of immune cell recruitment during the initial stage of infection. These findings suggest a new role of Mincle signaling on neutrophils during anti-mycobacterial responses.
in vivo neutrophil depletion, Immunofluorescence
Edelson, B. T., et al. (2011). "CD8alpha(+) dendritic cells are an obligate cellular entry point for productive infection by Listeria monocytogenes" Immunity 35(2): 236-248. PubMed
CD8alpha(+) dendritic cells (DCs) prime cytotoxic T lymphocytes during viral infections and produce interleukin-12 in response to pathogens. Although the loss of CD8alpha(+) DCs in Batf3(-/-) mice increases their susceptibility to several pathogens, we observed that Batf3(-/-) mice exhibited enhanced resistance to the intracellular bacterium Listeria monocytogenes. In wild-type mice, Listeria organisms, initially located in the splenic marginal zone, migrated to the periarteriolar lymphoid sheath (PALS) where they grew exponentially and induced widespread lymphocyte apoptosis. In Batf3(-/-) mice, however, Listeria organisms remain trapped in the marginal zone, failed to traffic into the PALS, and were rapidly cleared by phagocytes. In addition, Batf3(-/-) mice, which lacked the normal population of hepatic CD103(+) peripheral DCs, also showed protection from liver infection. These results suggest that Batf3-dependent CD8alpha(+) and CD103(+) DCs provide initial cellular entry points within the reticuloendothelial system by which Listeria establishes productive infection.
in vivo neutrophil depletion
Carr, K. D., et al. (2011). "Specific depletion reveals a novel role for neutrophil-mediated protection in the liver during Listeria monocytogenes infection" Eur J Immunol 41(9): 2666-2676. PubMed
Previous studies have suggested that neutrophils are required for resistance during infection with multiple pathogenic microorganisms. However, the depleting antibody used in those studies binds to both Ly6G and Ly6C (anti-Gr-1; clone RB6-8C5). This antibody has been shown to deplete not only neutrophils but also monocytes and a subset of CD8(+) T cells. Recently, an antibody against Ly6G, which specifically depletes neutrophils, was characterized. In the present study, neutrophils are depleted using the antibody against Ly6G during infection with the intracellular bacterium Listeria monocytogenes (LM). Our data show that neutrophil-depleted mice are much less susceptible to infection than mice depleted with anti-Gr-1. Although neutrophils are required for clearance of LM, their importance is more pronounced in the liver and during a high-dose bacterial challenge. Furthermore, we demonstrate that the protection mediated by neutrophils is due to the production of TNF-alpha, but not IFN-gamma. Additionally, neutrophils are not required for the recruitment of monocytes or the generation of adaptive T-cell responses during LM infection. This study highlights the importance of neutrophils during LM infection, and indicate that depletion of neutrophils is less detrimental to the host than depletion of all Gr-1-expressing cell populations.
in vivo neutrophil depletion, Flow Cytometry
Bamboat, Z. M., et al. (2010). "Conventional DCs reduce liver ischemia/reperfusion injury in mice via IL-10 secretion" J Clin Invest 120(2): 559-569. PubMed
TLRs are recognized as promoters of tissue damage, even in the absence of pathogens. TLR binding to damage-associated molecular patterns (DAMPs) released by injured host cells unleashes an inflammatory cascade that amplifies tissue destruction. However, whether TLRs possess the reciprocal ability to curtail the extent of sterile inflammation is uncertain. Here, we investigated this possibility in mice by studying the role of conventional DCs (cDCs) in liver ischemia/reperfusion (I/R) injury, a model of sterile inflammation. Targeted depletion of mouse cDCs increased liver injury after I/R, as assessed by serum alanine aminotransferase and histologic analysis. In vitro, we identified hepatocyte DNA as an endogenous ligand to TLR9 that promoted cDCs to secrete IL-10. In vivo, cDC production of IL-10 required TLR9 and reduced liver injury. In addition, we found that inflammatory monocytes recruited to the liver via chemokine receptor 2 were downstream targets of cDC IL-10. IL-10 from cDCs reduced production of TNF, IL-6, and ROS by inflammatory monocytes. Our results implicate inflammatory monocytes as mediators of liver I/R injury and reveal that cDCs respond to DAMPS during sterile inflammation, providing the host with protection from progressive tissue damage.