InVivoMAb anti-human PD-L1 (B7-H1)
Product Details
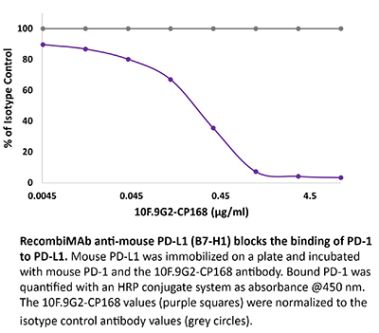
The 29E.2A3 monoclonal antibody reacts with human PD-L1 (programmed death ligand 1) also known as B7-H1 or CD274. PD-L1 is a 40 kDa type I transmembrane protein that belongs to the B7 family of the Ig superfamily. PD-L1 is expressed on T lymphocytes, B lymphocytes, NK cells, dendritic cells, as well as IFNγ stimulated monocytes, epithelial cells and endothelial cells. PD-L1 binds to its receptor, PD-1, found on CD4 and CD8 thymocytes as well as activated T and B lymphocytes and myeloid cells. Engagement of PD-L1 with PD-1 leads to inhibition of TCR-mediated T cell proliferation and cytokine production. PD-L1 is thought to play an important role in tumor immune evasion. Induced PD-L1 expression is common in many tumors and results in increased resistance of tumor cells to CD8 T cell mediated lysis. The 29E.2A3 antibody has been shown to block the binding of PD-1-Ig to PD-L1.Specifications
| Isotype | Mouse IgG2b, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb mouse IgG2b isotype control, unknown specificity |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Full length human PD-L1 |
| Reported Applications |
in vitro PD-L1 blockade Functional assays Immunohistochemistry (frozen) Flow cytometry |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
<2EU/mg (<0.002EU/μg) Determined by LAL gel clotting assay |
| Purity |
>95% Determined by SDS-PAGE |
| Sterility | 0.2 μM filtered |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_2687808 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
Recommended Products
-

Recommended Isotype Control(s)
InVivoMAb mouse IgG2b isotype control, unknown specificity
-

Recommended Dilution Buffer
InVivoPure pH 7.0 Dilution Buffer
Functional Assays
Sivakumar, R., et al. (2019). "Organotypic tumor slice cultures provide a versatile platform for immuno-oncology and drug discovery" Oncoimmunology 8(12): e1670019. PubMed
Organotypic tumor slices represent a physiologically-relevant culture system for studying the tumor microenvironment. Systematic characterization of the tumor slice culture system will enable its effective application for translational research. Here, using flow cytometry-based immunophenotyping, we performed a comprehensive characterization of the immune cell composition in organotypic tumor slices prepared from four syngeneic mouse tumor models and a human liver tumor. We found that the immune cell compositions of organotypic tumor slices prepared on the same day as the tumor cores were harvested are similar. Differences were primarily observed in the lymphocyte population of a clinical hepatocellular carcinoma case. Viable populations of immune cells persisted in the tumor slices for 7 days. Despite some changes in the immune cell populations, we showed the utility of mouse tumor slices for assessing responses to immune-modulatory agents. Further, we demonstrated the ability to use patient-derived xenograft tumor slices for assessing responses to targeted and cytotoxic drugs. Overall, tumor slices provide a broadly useful platform for studying the tumor microenvironment and evaluating the preclinical efficacy of cancer therapeutics.
in vitro PD-L1 blockade
Broos, K., et al. (2019). "Single Domain Antibody-Mediated Blockade of Programmed Death-Ligand 1 on Dendritic Cells Enhances CD8 T-cell Activation and Cytokine Production" Vaccines (Basel) 7(3). PubMed
Dendritic cell [DC] vaccines can induce durable clinical responses, at least in a fraction of previously treated, late stage cancer patients. Several preclinical studies suggest that shielding programmed death-ligand 1 [PD-L1] on the DC surface may be an attractive strategy to extend such clinical benefits to a larger patient population. In this study, we evaluated the use of single domain antibody [sdAb] K2, a high affinity, antagonistic, PD-L1 specific sdAb, for its ability to enhance DC mediated T-cell activation and benchmarked it against the use of the monoclonal antibodies [mAbs], MIH1, 29E.2A3 and avelumab. Similar to mAbs, sdAb K2 enhanced antigen-specific T-cell receptor signaling in PD-1 positive (PD-1(pos)) reporter cells activated by DCs. We further showed that the activation and function of antigen-specific CD8 positive (CD8(pos)) T cells, activated by DCs, was enhanced by inclusion of sdAb K2, but not mAbs. The failure of mAbs to enhance T-cell activation might be explained by their low efficacy to bind PD-L1 on DCs when compared to binding of PD-L1 on non-immune cells, whereas sdAb K2 shows high binding to PD-L1 on immune as well as non-immune cells. These data provide a rationale for the inclusion of sdAb K2 in DC-based immunotherapy strategies.
in vitro PD-L1 blockade
Porichis, F., et al. (2014). "Differential impact of PD-1 and/or interleukin-10 blockade on HIV-1-specific CD4 T cell and antigen-presenting cell functions" J Virol 88(5): 2508-2518. PubMed
Antigen persistence in chronic infections and cancer upregulates inhibitory networks, such as the PD-1 and interleukin-10 (IL-10) pathways, that impair immunity and lead to disease progression. These pathways are attractive targets for immunotherapy, as demonstrated by recent clinical trials of PD-1/PD-L1 blockade in cancer patients. However, in HIV-1 infection not all subjects respond to inhibition of either pathway and the mechanistic interactions between these two networks remain to be better defined. Here we demonstrate that in vitro blockade of PD-L1 and/or IL-10Ralpha results in markedly different profiles of HIV-1-specific CD4 T cell restoration. Whereas PD-L1 blockade leads to balanced increase in gamma interferon (IFN-gamma), IL-2, and IL-13 secretion, IL-10Ralpha blockade preferentially restores IFN-gamma production. In viremic subjects, combined PD-L1/IL-10Ralpha blockade results in a striking 10-fold increase in IFN-gamma secretion by HIV-1-specific CD4 T cells that is not observed in subjects with spontaneous (elite controllers) or therapy-induced control of viral replication. In contrast to the dramatic increase in IFN-gamma production, concurrent blockade has a marginal additive effect on IL-2 production, IL-13 secretion, and HIV-1-specific CD4 T cell proliferation. IFN-gamma produced by Thelper cells upregulates PD-L1, HLA I/II, and IL-12 expression by monocytes. The effect of combined blockade on IFN-gamma was dependent on reciprocal reinforcement through IL-12. These studies provide crucial information on the different immunoregulatory qualities of PD-1 and IL-10 in progressive disease and link exhausted virus-specific CD4 T cells and monocytes in the regulation of IFN-gamma and IL-12 secretion. IMPORTANCE: Infection with HIV results in most people in uncontrolled viral replication and progressive weakening of the body defenses. In the absence of antiviral therapy, this process results in clinical disease, or AIDS. An important reason why HIV continues to multiply is that a population of white blood cells called CD4 T cells that targets the virus fails to work properly. At least part of this impairment is under the control of inhibitory mechanisms that can be blocked to improve the function of these CD4 T cells. In this report, we show that blocking one or two of the molecules involved, called PD-1 and IL-10, has different effects on the individual functions of these cells and that one is strongly improved. We investigate how these effects are caused by interactions between CD4 T cells and antigen-presenting cells. These observations can have implications for new therapeutic approaches in HIV infection.
in vitro PD-L1 blockade
Hegde, S., et al. (2011). "Human NKT cells direct the differentiation of myeloid APCs that regulate T cell responses via expression of programmed cell death ligands" J Autoimmun 37(1): 28-38. PubMed
NKT cells are innate lymphocytes that can recognize self or foreign lipids presented by CD1d molecules. NKT cells have been shown to inhibit the development of autoimmunity in murine model systems, however, the pathways by which they foster immune tolerance remain poorly understood. Here we show that autoreactive human NKT cells stimulate monocytes to differentiate into myeloid APCs that have a regulatory phenotype characterized by poor conjugate formation with T cells. The NKT cell instructed myeloid APCs show elevated expression of the inhibitory ligand PD-L2, and blocking PD-L1 and PD-L2 during interactions of the APCs with T cells results in improved cluster formation and significantly increased T cell proliferative responses. The elevated expression of PD-L molecules on NKT-instructed APCs appears to result from exposure to extracellular ATP that is produced during NKT-monocyte interactions, and blocking purinergic signaling during monocyte differentiation results in APCs that form clusters with T cells and stimulate their proliferation. Finally, we show that human monocytes and NKT cells that are injected into immunodeficient mice co-localize together in spleen and liver, and after 3 days in vivo in the presence of NKT cells a fraction of the myeloid cells have upregulated markers associated with differentiation into professional APCs. These results suggest that autoreactive human NKT cells may promote tolerance by inducing the differentiation of regulatory myeloid APCs that limit T cell proliferation through expression of PD-L molecules.
in vitro PD-L1 blockade
Nakamoto, N., et al. (2009). "Synergistic reversal of intrahepatic HCV-specific CD8 T cell exhaustion by combined PD-1/CTLA-4 blockade" PLoS Pathog 5(2): e1000313. PubMed
Viral persistence is associated with hierarchical antiviral CD8 T cell exhaustion with increased programmed death-1 (PD-1) expression. In HCV persistence, HCV-specific CD8 T cells from the liver (the site of viral replication) display increased PD-1 expression and a profound functional impairment that is not reversed by PD-1 blockade alone. Here, we report that the inhibitory receptor cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) is preferentially upregulated in PD-1(+) T cells from the liver but not blood of chronically HCV-infected patients. PD-1/CTLA-4 co-expression in intrahepatic T cells was associated with a profound HCV-specific effector dysfunction that was synergistically reversed by combined PD-1/CTLA-4 blockade in vitro, but not by blocking PD-1 or CTLA-4 alone. A similar effect was observed in circulating HCV-specific CD8 T cells with increased PD-1/CTLA-4 co-expression during acute hepatitis C. The functional response to combined blockade was directly associated with CTLA-4 expression, lost with CD28-depletion and CD4-independent (including CD4(+)FoxP3(+) Tregs). We conclude that PD-1 and CTLA-4 pathways both contribute to virus-specific T cell exhaustion at the site of viral replication by a redundant mechanism that requires combined PD-1/CTLA-4 blockade to reverse. These findings provide new insights into the mechanisms of virus-specific T cell dysfunction, and suggest that the synergistic effect by combined inhibitory receptor blockade might have a therapeutic application against chronic viral infection in vivo, provided that it does not induce autoimmunity.
in vitro PD-L1 blockade
Cai, G., et al. (2004). "PD-1 ligands, negative regulators for activation of naive, memory, and recently activated human CD4+ T cells" Cell Immunol 230(2): 89-98. PubMed
We examined the role of the PD-1 pathway on the activation of naive, memory, and recently activated human CD4+ T cells to test whether they responded differently. PD-1 ligand blockade modestly enhanced the percentage of responding T cells and production of IFN-gamma in a primary response to myelin basic protein (MBP) in normal donors. PD-1 ligand blockade strongly enhanced proliferation and cytokine production by memory or recently activated T cells (tetanus toxoid and MBP). Blockade of PD-L1 alone had more effect than PD-L2, consistent with its higher expression on ex vivo dendritic cells; furthermore, anti-PD-L1 plus anti-PD-L2 resulted in the greatest enhancement. Moreover, PD-L1-Ig inhibited anti-CD3 induced activation of naive, memory, and recently activated CD4+ T cells. Together, our data demonstrated PD-1 functioned as a negative regulatory pathway on naive T cells during a primary response, and more potently, on memory or recently activated T cells during a secondary response.
Functional Assays, Flow Cytometry, Immunohistochemistry (frozen)
Brown, J. A., et al. (2003). "Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production" J Immunol 170(3): 1257-1266. PubMed
Programmed death-1 ligand (PD-L)1 and PD-L2 are ligands for programmed death-1 (PD-1), a member of the CD28/CTLA4 family expressed on activated lymphoid cells. PD-1 contains an immunoreceptor tyrosine-based inhibitory motif and mice deficient in PD-1 develop autoimmune disorders suggesting a defect in peripheral tolerance. Human PD-L1 and PD-L2 are expressed on immature dendritic cells (iDC) and mature dendritic cells (mDC), IFN-gamma-treated monocytes, and follicular dendritic cells. Using mAbs, we show that blockade of PD-L2 on dendritic cells results in enhanced T cell proliferation and cytokine production, including that of IFN-gamma and IL-10, while blockade of PD-L1 results in similar, more modest, effects. Blockade of both PD-L1 and PD-L2 showed an additive effect. Both whole mAb and Fab enhanced T cell activation, showing that PD-L1 and PD-L2 function to inhibit T cell activation. Enhancement of T cell activation was most pronounced with weak APC, such as iDCs and IL-10-pretreated mDCs, and less pronounced with strong APC such as mDCs. These data are consistent with the hypothesis that iDC have a balance of stimulatory vs inhibitory molecules that favors inhibition, and indicate that PD-L1 and PD-L2 contribute to the poor stimulatory capacity of iDC. PD-L1 expression differs from PD-L2 in that PD-L1 is expressed on activated T cells, placental trophoblasts, myocardial endothelium, and cortical thymic epithelial cells. In contrast, PD-L2 is expressed on placental endothelium and medullary thymic epithelial cells. PD-L1 is also highly expressed on most carcinomas but minimally expressed on adjacent normal tissue suggesting a role in attenuating antitumor immune responses.