InVivoMAb anti-human CD4
Product Details
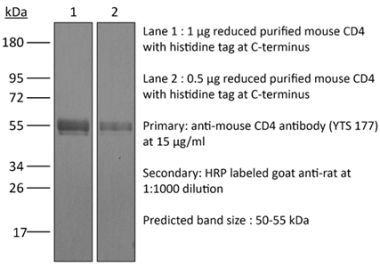
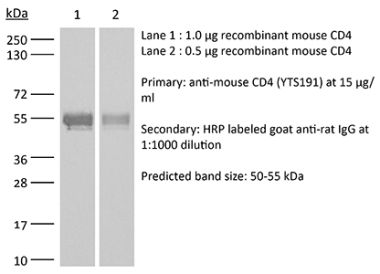
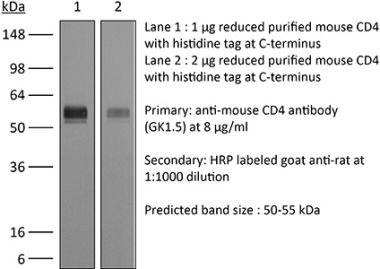
The RPA-T4 monoclonal antibody reacts with the human CD4. The CD4 antigen is a 55 kDa cell surface type I membrane glycoprotein belonging to the immunoglobulin superfamily. CD4 acts as a co-receptor which in cooperation with the T cell receptor (TCR) interacts with class II MHC molecules displayed by antigen presenting cells (APC). CD4 is expressed by most thymocytes and helper T cells, a subset of NK-T cells and weakly by dendritic cells and macrophages. CD4 plays an important role in the development of T cells and is required for mature T cells to function optimally. The RPA-T4 antibody is reported to bind to the D1 domain of CD4 and does not block the binding of the OKT-4 antibody. Additionally, RPA-T4 has been shown to block the binding of HIV gp120 protein to CD4 and inhibit CD4 T cell activation in vitro.Specifications
| Isotype | Mouse IgG1, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb mouse IgG1 isotype control, unknown specificity |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Not available or unknown |
| Reported Applications |
in vitro CD4 blockade in vitro blocking of CD4+ T cell activation Immunofluorescence Immunohistochemistry (frozen) Flow cytometry |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
<2EU/mg (<0.002EU/μg) Determined by LAL gel clotting assay |
| Purity |
>95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_2687811 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
Recommended Products
-

Recommended Isotype Control(s)
InVivoMAb mouse IgG1 isotype control, unknown specificity
-

Recommended Dilution Buffer
InVivoPure pH 7.0 Dilution Buffer
Immunofluorescence
Moolla, N., et al. (2016). "Thioredoxin (Trx1) regulates CD4 membrane domain localization and is required for efficient CD4-dependent HIV-1 entry" Biochim Biophys Acta 1860(9): 1854-1863. PubMed
BACKGROUND: CD4 is a glycoprotein expressed on the surfaces of certain immune cells. On lymphocytes, an important function of CD4 is to co-engage Major Histocompatibility Complex (MHC) molecules with the T Cell Receptor (TCR), a process that is essential for antigen-specific activation of T cells. CD4 localizes dynamically into distinct membrane microdomains, an important feature of its immunoregulatory function that has also been shown to influence the efficiency of HIV replication. However, the mechanism by which CD4 localization is regulated and the biological significance of this is incompletely understood. METHODS: In this study, we used confocal microscopy, density-gradient centrifugation and flow cytometry to analyze dynamic redox-dependent effects on CD4 membrane domain localization. RESULTS: Blocking cell surface redox exchanges with both a membrane-impermeable sulfhydryl blocker (DTNB) and specific antibody inhibitors of Thioredoxin-1 (Trx1) induces translocation of CD4 into detergent-resistant membrane domains (DRM). In contrast, Trx1 inactivation does not change the localization of the chemokine receptor CCR5, suggesting that this effect is targeted. Moreover, DTNB treatment and Trx1 depletion coincide with strong inhibition of CD4-dependent HIV entry, but only moderate reductions in the infectivity of a CD4-independent HIV pseudovirion. CONCLUSIONS: Changes in the extracellular redox environment, potentially mediated by allosteric consequences of functional disulfide bond oxidoreduction, may represent a signal for translocation of CD4 into DRM clusters, and this sequestration, another potential mechanism by which the anti-HIV effects of cell surface oxidoreductase inhibition are exerted. GENERAL SIGNIFICANCE: Extracellular redox conditions may regulate CD4 function by potentiating changes in its membrane domain localization.
in vitro blocking of T cell activation
Mayer, C. T., et al. (2013). "CD4 blockade directly inhibits mouse and human CD4(+) T cell functions independent of Foxp3(+) Tregs" J Autoimmun 47: 73-82. PubMed
CD4(+) helper T cells orchestrate protective immunity against pathogens, yet can also induce undesired pathologies including allergies, transplant rejection and autoimmunity. Non-depleting CD4-specific antibodies such as clone YTS177.9 were found to promote long-lasting T cell tolerance in animal models. Thus, CD4 blockade could represent a promising therapeutic approach for human autoimmune diseases. However, the mechanisms underlying anti-CD4-induced tolerance are incompletely resolved. Particularly, multiple immune cells express CD4 including Foxp3(+) regulatory T cells (Tregs) and dendritic cells (DCs), both controlling the activation of CD4(+)Foxp3(-) helper T cells. Utilizing mixed leukocyte reactions (MLRs) reflecting physiological interactions between T cells and DCs, we report that anti-CD4 treatment inhibits CD4(+)Foxp3(-) T cell proliferation in an IL-2-independent fashion. Notably, YTS177.9 binding induces a rapid internalization of CD4 on both CD4(+)Foxp3(-) T cells and Foxp3(+) Tregs. However, no expansion or activation of immunosuppressive CD4(+)Foxp3(+) Tregs was observed following anti-CD4 treatment. Additionally, cytokine production, maturation and T cell priming capacity of DCs are not affected by anti-CD4 exposure. In line with these data, the selective ablation of Foxp3(+) Tregs from MLRs by the use of diphtheria toxin (DT)-treated bacterial artificial chromosome (BAC)-transgenic DEREG mice completely fails to abrogate the suppressive activity of multiple anti-CD4 antibodies. Instead, tolerization is associated with the defective expression of various co-stimulatory receptors including OX40 and CD30, suggesting altered signaling through the TCR complex. Consistent with our findings in mice, anti-CD4 treatment renders human CD4(+) T cells tolerant in the absence of Tregs. Thus, our results establish that anti-CD4 antibodies can directly tolerize pathogenic CD4(+)Foxp3(-) helper T cells. This has important implications for the treatment of human inflammatory diseases.
in vitro CD4 blockade
Toma, J., et al. (2011). "Loss of asparagine-linked glycosylation sites in variable region 5 of human immunodeficiency virus type 1 envelope is associated with resistance to CD4 antibody ibalizumab" J Virol 85(8): 3872-3880. PubMed
Ibalizumab (formerly TNX-355) is a first-in-class, monoclonal antibody inhibitor of CD4-mediated human immunodeficiency type 1 (HIV-1) entry. Multiple clinical trials with HIV-infected patients have demonstrated the antiviral activity, safety, and tolerability of ibalizumab treatment. A 9-week phase Ib study adding ibalizumab monotherapy to failing drug regimens led to transient reductions in HIV viral loads and the evolution of HIV-1 variants with reduced susceptibility to ibalizumab. This report characterizes these variants by comparing the phenotypic susceptibilities and envelope (env) sequences of (i) paired baseline and on-treatment virus populations, (ii) individual env clones from selected paired samples, and (iii) env clones containing site-directed mutations. Viruses with reduced susceptibility to ibalizumab were found to exhibit reduced susceptibility to the anti-CD4 antibody RPA-T4. Conversely, susceptibility to soluble CD4, which targets the HIV-1 gp120 envelope protein, was enhanced. No changes in susceptibility to the fusion inhibitor enfuvirtide or the CCR5 antagonist maraviroc were observed. Functionally, viruses with reduced ibalizumab susceptibility also displayed high levels of infectivity relative to those of paired baseline viruses. Individual env clones exhibiting reduced ibalizumab susceptibility contained multiple amino acid changes in different regions relative to the paired baseline clones. In particular, clones with reduced susceptibility to ibalizumab contained fewer potential asparagine-linked glycosylation sites (PNGSs) in variable region 5 (V5) than did paired ibalizumab-susceptible clones. The reduction in ibalizumab susceptibility due to the loss of V5 PNGSs was confirmed by site-directed mutagenesis. Taken together, these findings provide important insights into resistance to this new class of antiretroviral drug.
Immunohistochemistry (frozen)
Mack, C. L., et al. (2004). "Biliary atresia is associated with CD4+ Th1 cell-mediated portal tract inflammation" Pediatr Res 56(1): 79-87. PubMed
A proposed mechanism in the pathogenesis of biliary atresia involves an initial virus-induced, progressive T cell-mediated inflammatory obliteration of bile ducts. The aim of this study was to characterize the inflammatory environment present within the liver of infants with biliary atresia to gain insight into the role of a primary immune-mediated process versus a nonspecific secondary response to biliary obstruction. Frozen liver tissue obtained from patients with biliary atresia, neonatal giant cell hepatitis, total parenteral nutrition (TPN)-related cholestasis, choledochal cysts, and normal control subjects was used for fluorescent immunohistochemistry studies of cellular infiltrates, cytokine mRNA expression, and in situ hybridization for localization of cytokine-producing cells. Immunohistochemistry revealed increases in CD8(+) and CD4(+) T cells and Kupffer cells (CD68(+)) in the portal tracts of biliary atresia. Reverse transcription-PCR analysis of biliary atresia tissue showed a Th1-type cytokine profile with expression of IL-2, interferon-gamma, tumor necrosis factor-alpha, and IL-12. This profile was not seen in normal, neonatal hepatitis or choledochal cyst livers but was present in TPN-related cholestasis. In situ hybridization revealed that the Th1 cytokine-producing cells were located in the portal tracts in biliary atresia and in the parenchyma of TPN-related cholestasis. A distinctive portal tract inflammatory environment is present in biliary atresia, involving CD4(+) Th1 cell-mediated immunity. The absence of similar inflammation in other pediatric cholestatic conditions suggests that the portal tract inflammation in biliary atresia is not a secondary response to cholestasis but rather indicates a specific immune response involved in the pathogenesis of biliary atresia.
in vitro CD4 blockade, Flow Cytometry
Geijtenbeek, T. B., et al. (2000). "DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells" Cell 100(5): 587-597. PubMed
Dendritic cells (DC) capture microorganisms that enter peripheral mucosal tissues and then migrate to secondary lymphoid organs, where they present these in antigenic form to resting T cells and thus initiate adaptive immune responses. Here, we describe the properties of a DC-specific C-type lectin, DC-SIGN, that is highly expressed on DC present in mucosal tissues and binds to the HIV-1 envelope glycoprotein gp120. DC-SIGN does not function as a receptor for viral entry into DC but instead promotes efficient infection in trans of cells that express CD4 and chemokine receptors. We propose that DC-SIGN efficiently captures HIV-1 in the periphery and facilitates its transport to secondary lymphoid organs rich in T cells, to enhance infection in trans of these target cells.
in vitro CD4 blockade
Moir, S., et al. (1999). "CD40-Mediated induction of CD4 and CXCR4 on B lymphocytes correlates with restricted susceptibility to human immunodeficiency virus type 1 infection: potential role of B lymphocytes as a viral reservoir" J Virol 73(10): 7972-7980. PubMed
Human immunodeficiency virus type 1 (HIV-1) replicates primarily in lymphoid tissues where it has ready access to activated immune competent cells. We used one of the major pathways of immune activation, namely, CD40-CD40L interactions, to study the infectability of B lymphocytes isolated from peripheral blood mononuclear cells. Highly enriched populations of B lymphocytes generated in the presence of interleukin-4 and oligomeric soluble CD40L upregulated costimulatory and activation markers, as well as HIV-1 receptors CD4 and CXCR4, but not CCR5. By using single-round competent luciferase viruses complemented with either amphotropic or HIV-derived envelopes, we found a direct correlation between upregulation of HIV-1 receptors and the susceptibility of the B lymphocytes to infection with dual-tropic and T-tropic strains of HIV-1; in contrast, cells were resistant to M-tropic strains of HIV-1. HIV-1 envelope-mediated infection was completely abolished with either an anti-CD4 monoclonal antibody or a peptide known to directly block CXCR4 usage and partially blocked with stromal cell-derived factor 1, all of which had no effect on the entry of virus pseudotyped with amphotropic envelope. Full virus replication kinetics confirmed that infection depends on CXCR4 usage. Furthermore, productive cycles of virus replication occurred rapidly yet under most conditions, without the appearance of syncytia. Thus, an activated immunological environment may induce the expression of HIV-1 receptors on B lymphocytes, priming them for infection with selective strains of HIV-1 and allowing them to serve as a potential viral reservoir.